Jump to
S1C2
Vibrational Frequencies calculated at MP3=FULL/cc-pVDZ
Geometric Data calculated at MP3=FULL/cc-pVDZ
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at MP3=FULL/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -115.603103 |
| Energy at 298.15K | -115.604854 |
| HF Energy | -115.202210 |
| Nuclear repulsion energy | 57.372045 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at MP3=FULL/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3355 |
3155 |
0.54 |
|
|
|
| 2 |
A' |
3134 |
2947 |
51.43 |
|
|
|
| 3 |
A' |
1687 |
1586 |
0.26 |
|
|
|
| 4 |
A' |
1261 |
1185 |
9.72 |
|
|
|
| 5 |
A' |
1019 |
958 |
16.87 |
|
|
|
| 6 |
A' |
931 |
876 |
4.53 |
|
|
|
| 7 |
A' |
613 |
577 |
63.63 |
|
|
|
| 8 |
A" |
4591 |
4317 |
61197.95 |
|
|
|
| 9 |
A" |
2993 |
2814 |
4994.13 |
|
|
|
| 10 |
A" |
1022 |
961 |
11.89 |
|
|
|
| 11 |
A" |
985 |
926 |
0.02 |
|
|
|
| 12 |
A" |
873 |
821 |
0.14 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 11231.9 cm
-1
Scaled (by 0.9403) Zero Point Vibrational Energy (zpe) 10561.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at MP3=FULL/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.038 |
0.898 |
0.000 |
| H2 |
0.754 |
1.664 |
0.000 |
| C3 |
-0.038 |
-0.423 |
0.664 |
| C4 |
-0.038 |
-0.423 |
-0.664 |
| H5 |
-0.037 |
-0.992 |
1.590 |
| H6 |
-0.037 |
-0.992 |
-1.590 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
C3 |
C4 |
H5 |
H6 |
| C1 | | 1.1015 | 1.4784 | 1.4784 | 2.4697 | 2.4697 |
H2 | 1.1015 | | 2.3282 | 2.3282 | 3.1945 | 3.1945 | C3 | 1.4784 | 2.3282 | | 1.3278 | 1.0869 | 2.3246 | C4 | 1.4784 | 2.3282 | 1.3278 | | 2.3246 | 1.0869 | H5 | 2.4697 | 3.1945 | 1.0869 | 2.3246 | | 3.1800 | H6 | 2.4697 | 3.1945 | 2.3246 | 1.0869 | 3.1800 | |
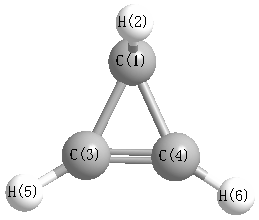 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
C4 |
63.315 |
|
C1 |
C3 |
H5 |
148.251 |
| C1 |
C4 |
C3 |
63.315 |
|
C1 |
C4 |
H6 |
148.251 |
| H2 |
C1 |
C3 |
128.374 |
|
H2 |
C1 |
C4 |
128.374 |
| C3 |
C1 |
C4 |
53.371 |
|
C3 |
C4 |
H6 |
148.435 |
| C4 |
C3 |
H5 |
148.435 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability