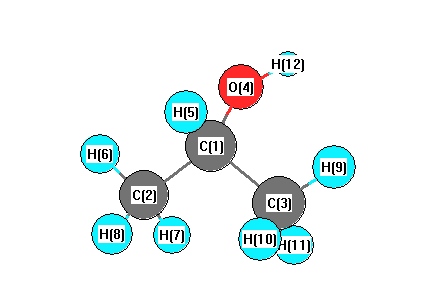
Vibrational Frequencies calculated at B3LYP/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3817 |
3684 |
10.82 |
|
|
|
| 2 |
A |
3122 |
3013 |
24.98 |
|
|
|
| 3 |
A |
3116 |
3007 |
49.06 |
|
|
|
| 4 |
A |
3108 |
2999 |
4.77 |
|
|
|
| 5 |
A |
3091 |
2982 |
33.08 |
|
|
|
| 6 |
A |
3041 |
2935 |
11.73 |
|
|
|
| 7 |
A |
3025 |
2919 |
22.15 |
|
|
|
| 8 |
A |
2951 |
2847 |
61.49 |
|
|
|
| 9 |
A |
1510 |
1457 |
3.81 |
|
|
|
| 10 |
A |
1497 |
1445 |
3.64 |
|
|
|
| 11 |
A |
1488 |
1435 |
1.69 |
|
|
|
| 12 |
A |
1482 |
1430 |
0.74 |
|
|
|
| 13 |
A |
1430 |
1380 |
19.05 |
|
|
|
| 14 |
A |
1411 |
1361 |
14.01 |
|
|
|
| 15 |
A |
1391 |
1343 |
1.47 |
|
|
|
| 16 |
A |
1377 |
1329 |
15.72 |
|
|
|
| 17 |
A |
1283 |
1238 |
50.91 |
|
|
|
| 18 |
A |
1188 |
1146 |
32.09 |
|
|
|
| 19 |
A |
1165 |
1124 |
20.87 |
|
|
|
| 20 |
A |
1086 |
1048 |
20.91 |
|
|
|
| 21 |
A |
969 |
935 |
41.49 |
|
|
|
| 22 |
A |
947 |
914 |
1.78 |
|
|
|
| 23 |
A |
925 |
892 |
0.15 |
|
|
|
| 24 |
A |
823 |
794 |
3.02 |
|
|
|
| 25 |
A |
478 |
462 |
5.66 |
|
|
|
| 26 |
A |
411 |
397 |
8.72 |
|
|
|
| 27 |
A |
357 |
344 |
1.94 |
|
|
|
| 28 |
A |
301 |
291 |
97.91 |
|
|
|
| 29 |
A |
261 |
252 |
2.08 |
|
|
|
| 30 |
A |
216 |
209 |
4.29 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23631.4 cm
-1
Scaled (by 0.965) Zero Point Vibrational Energy (zpe) 22804.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.156 |
|
|
|
| 2 |
C |
-0.390 |
|
|
|
| 3 |
C |
-0.364 |
|
|
|
| 4 |
O |
-0.432 |
|
|
|
| 5 |
H |
0.062 |
|
|
|
| 6 |
H |
0.102 |
|
|
|
| 7 |
H |
0.123 |
|
|
|
| 8 |
H |
0.114 |
|
|
|
| 9 |
H |
0.123 |
|
|
|
| 10 |
H |
0.110 |
|
|
|
| 11 |
H |
0.120 |
|
|
|
| 12 |
H |
0.278 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.080 |
-0.606 |
0.708 |
1.427 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
89.157 |
| (<r2>)1/2 |
9.442 |