Vibrational Frequencies calculated at TPSSh/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3201 |
3071 |
3.41 |
|
|
|
| 2 |
A' |
3178 |
3049 |
9.62 |
|
|
|
| 3 |
A' |
3132 |
3005 |
30.21 |
|
|
|
| 4 |
A' |
3055 |
2931 |
20.27 |
|
|
|
| 5 |
A' |
3024 |
2901 |
20.78 |
|
|
|
| 6 |
A' |
2315 |
2221 |
25.23 |
|
|
|
| 7 |
A' |
1701 |
1632 |
26.27 |
|
|
|
| 8 |
A' |
1548 |
1485 |
4.43 |
|
|
|
| 9 |
A' |
1503 |
1442 |
9.02 |
|
|
|
| 10 |
A' |
1450 |
1391 |
3.83 |
|
|
|
| 11 |
A' |
1397 |
1341 |
11.71 |
|
|
|
| 12 |
A' |
1349 |
1295 |
0.93 |
|
|
|
| 13 |
A' |
1309 |
1256 |
1.93 |
|
|
|
| 14 |
A' |
1119 |
1074 |
1.91 |
|
|
|
| 15 |
A' |
1072 |
1029 |
4.82 |
|
|
|
| 16 |
A' |
993 |
952 |
1.74 |
|
|
|
| 17 |
A' |
900 |
863 |
3.50 |
|
|
|
| 18 |
A' |
574 |
551 |
0.35 |
|
|
|
| 19 |
A' |
503 |
483 |
0.04 |
|
|
|
| 20 |
A' |
255 |
244 |
1.27 |
|
|
|
| 21 |
A' |
144 |
138 |
4.02 |
|
|
|
| 22 |
A" |
3125 |
2998 |
33.55 |
|
|
|
| 23 |
A" |
3046 |
2923 |
13.48 |
|
|
|
| 24 |
A" |
1543 |
1480 |
6.82 |
|
|
|
| 25 |
A" |
1304 |
1251 |
0.20 |
|
|
|
| 26 |
A" |
1113 |
1068 |
1.21 |
|
|
|
| 27 |
A" |
1004 |
964 |
29.67 |
|
|
|
| 28 |
A" |
846 |
811 |
7.15 |
|
|
|
| 29 |
A" |
737 |
707 |
0.28 |
|
|
|
| 30 |
A" |
490 |
470 |
6.05 |
|
|
|
| 31 |
A" |
277 |
266 |
0.01 |
|
|
|
| 32 |
A" |
177 |
170 |
0.01 |
|
|
|
| 33 |
A" |
123 |
118 |
3.70 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23753.3 cm
-1
Scaled (by 0.9594) Zero Point Vibrational Energy (zpe) 22788.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
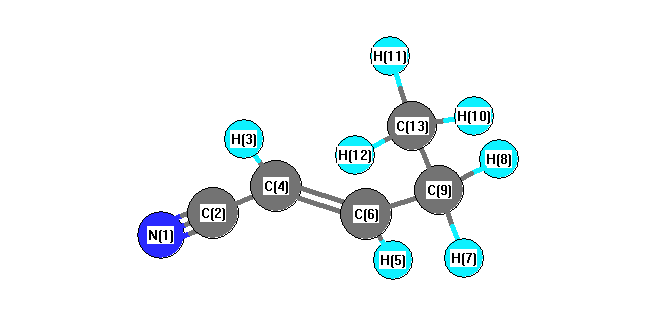
Charges from optimized geometry at TPSSh/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.500 |
|
|
|
| 2 |
C |
0.333 |
|
|
|
| 3 |
H |
0.201 |
|
|
|
| 4 |
C |
-0.177 |
|
|
|
| 5 |
H |
0.185 |
|
|
|
| 6 |
C |
-0.089 |
|
|
|
| 7 |
H |
0.183 |
|
|
|
| 8 |
H |
0.183 |
|
|
|
| 9 |
C |
-0.337 |
|
|
|
| 10 |
H |
0.173 |
|
|
|
| 11 |
H |
0.171 |
|
|
|
| 12 |
H |
0.171 |
|
|
|
| 13 |
C |
-0.498 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.111 |
-4.337 |
0.000 |
4.823 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.887 |
-1.927 |
0.000 |
| y |
-1.927 |
12.664 |
0.000 |
| z |
0.000 |
0.000 |
5.086 |
<r2> (average value of r
2) Å
2
| <r2> |
227.266 |
| (<r2>)1/2 |
15.075 |