Vibrational Frequencies calculated at TPSSh/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3219 |
3113 |
36.51 |
|
|
|
| 2 |
A' |
3138 |
3035 |
23.13 |
|
|
|
| 3 |
A' |
3125 |
3023 |
18.16 |
|
|
|
| 4 |
A' |
3086 |
2985 |
26.65 |
|
|
|
| 5 |
A' |
3015 |
2917 |
44.75 |
|
|
|
| 6 |
A' |
1511 |
1461 |
1.07 |
|
|
|
| 7 |
A' |
1493 |
1444 |
6.81 |
|
|
|
| 8 |
A' |
1417 |
1370 |
2.66 |
|
|
|
| 9 |
A' |
1383 |
1338 |
3.47 |
|
|
|
| 10 |
A' |
1230 |
1190 |
0.04 |
|
|
|
| 11 |
A' |
1206 |
1166 |
0.05 |
|
|
|
| 12 |
A' |
1038 |
1004 |
3.59 |
|
|
|
| 13 |
A' |
1000 |
967 |
10.79 |
|
|
|
| 14 |
A' |
931 |
900 |
2.10 |
|
|
|
| 15 |
A' |
791 |
766 |
5.98 |
|
|
|
| 16 |
A' |
761 |
736 |
0.88 |
|
|
|
| 17 |
A' |
354 |
342 |
0.21 |
|
|
|
| 18 |
A" |
3203 |
3099 |
0.36 |
|
|
|
| 19 |
A" |
3120 |
3018 |
33.65 |
|
|
|
| 20 |
A" |
3081 |
2980 |
38.75 |
|
|
|
| 21 |
A" |
1481 |
1433 |
1.53 |
|
|
|
| 22 |
A" |
1465 |
1417 |
1.65 |
|
|
|
| 23 |
A" |
1186 |
1147 |
0.58 |
|
|
|
| 24 |
A" |
1126 |
1089 |
0.00 |
|
|
|
| 25 |
A" |
1100 |
1064 |
3.02 |
|
|
|
| 26 |
A" |
1048 |
1013 |
0.96 |
|
|
|
| 27 |
A" |
875 |
846 |
8.78 |
|
|
|
| 28 |
A" |
802 |
776 |
3.55 |
|
|
|
| 29 |
A" |
335 |
324 |
0.49 |
|
|
|
| 30 |
A" |
223 |
216 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23870.5 cm
-1
Scaled (by 0.9673) Zero Point Vibrational Energy (zpe) 23089.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
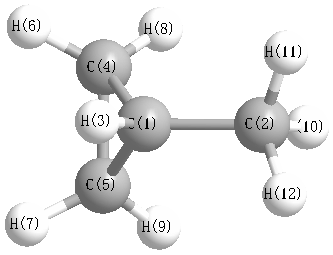
Charges from optimized geometry at TPSSh/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.679 |
|
|
|
| 2 |
C |
0.227 |
|
|
|
| 3 |
H |
-0.486 |
|
|
|
| 4 |
C |
0.937 |
|
|
|
| 5 |
C |
0.937 |
|
|
|
| 6 |
H |
-0.486 |
|
|
|
| 7 |
H |
-0.486 |
|
|
|
| 8 |
H |
-0.475 |
|
|
|
| 9 |
H |
-0.475 |
|
|
|
| 10 |
H |
-0.214 |
|
|
|
| 11 |
H |
-0.078 |
|
|
|
| 12 |
H |
-0.078 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.050 |
0.125 |
0.000 |
0.134 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.976 |
-0.566 |
0.000 |
| y |
-0.566 |
7.914 |
0.000 |
| z |
0.000 |
0.000 |
7.296 |
<r2> (average value of r
2) Å
2
| <r2> |
84.176 |
| (<r2>)1/2 |
9.175 |