Jump to
S1C2
Energy calculated at TPSSh/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -151.575325 |
| Energy at 298.15K | -151.577622 |
| HF Energy | -151.575325 |
| Nuclear repulsion energy | 36.636837 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3709 |
3587 |
9.06 |
112.33 |
0.11 |
0.19 |
| 2 |
A |
1423 |
1376 |
0.56 |
7.55 |
0.30 |
0.47 |
| 3 |
A |
929 |
898 |
0.60 |
13.92 |
0.20 |
0.34 |
| 4 |
A |
383 |
370 |
156.93 |
1.64 |
0.74 |
0.85 |
| 5 |
B |
3708 |
3587 |
40.39 |
30.27 |
0.75 |
0.86 |
| 6 |
B |
1312 |
1269 |
95.33 |
1.40 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 5730.8 cm
-1
Scaled (by 0.9673) Zero Point Vibrational Energy (zpe) 5543.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/aug-cc-pVDZ
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.729 |
-0.059 |
| O2 |
0.000 |
-0.729 |
-0.059 |
| H3 |
0.796 |
0.900 |
0.474 |
| H4 |
-0.796 |
-0.900 |
0.474 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
H3 |
H4 |
| O1 | | 1.4575 | 0.9733 | 1.8899 |
O2 | 1.4575 | | 1.8899 | 0.9733 | H3 | 0.9733 | 1.8899 | | 2.4036 | H4 | 1.8899 | 0.9733 | 2.4036 | |
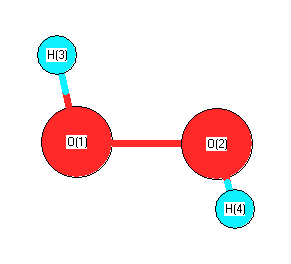 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
O2 |
H4 |
100.153 |
|
O2 |
O1 |
H3 |
100.153 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.201 |
|
|
|
| 2 |
O |
-0.201 |
|
|
|
| 3 |
H |
0.201 |
|
|
|
| 4 |
H |
0.201 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.742 |
1.742 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.060 |
0.254 |
0.000 |
| y |
0.254 |
2.812 |
0.000 |
| z |
0.000 |
0.000 |
1.883 |
<r2> (average value of r
2) Å
2
| <r2> |
18.845 |
| (<r2>)1/2 |
4.341 |
Jump to
S1C1
Energy calculated at TPSSh/aug-cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -151.573703 |
| Energy at 298.15K | |
| HF Energy | -151.573703 |
| Nuclear repulsion energy | 36.496520 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/aug-cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3747 |
3625 |
0.00 |
|
|
|
| 2 |
Ag |
1530 |
1480 |
0.00 |
|
|
|
| 3 |
Ag |
932 |
902 |
0.00 |
|
|
|
| 4 |
Au |
277i |
268i |
242.82 |
|
|
|
| 5 |
Bu |
3757 |
3634 |
81.17 |
|
|
|
| 6 |
Bu |
1232 |
1192 |
123.77 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5460.7 cm
-1
Scaled (by 0.9673) Zero Point Vibrational Energy (zpe) 5282.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/aug-cc-pVDZ
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.734 |
0.000 |
| O2 |
0.000 |
-0.734 |
0.000 |
| H3 |
0.961 |
0.878 |
0.000 |
| H4 |
-0.961 |
-0.878 |
0.000 |
Atom - Atom Distances (Å)
| |
O1 |
O2 |
H3 |
H4 |
| O1 | | 1.4682 | 0.9716 | 1.8766 |
O2 | 1.4682 | | 1.8766 | 0.9716 | H3 | 0.9716 | 1.8766 | | 2.6030 | H4 | 1.8766 | 0.9716 | 2.6030 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
O2 |
H4 |
98.505 |
|
O2 |
O1 |
H3 |
98.505 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/aug-cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.216 |
|
|
|
| 2 |
O |
-0.216 |
|
|
|
| 3 |
H |
0.216 |
|
|
|
| 4 |
H |
0.216 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.185 |
0.270 |
0.000 |
| y |
0.270 |
2.766 |
0.000 |
| z |
0.000 |
0.000 |
1.768 |
<r2> (average value of r
2) Å
2
| <r2> |
18.935 |
| (<r2>)1/2 |
4.351 |