Jump to
S1C2
S2C1
S2C2
Energy calculated at TPSSh/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -131.478317 |
| Energy at 298.15K | -131.477632 |
| HF Energy | -131.478317 |
| Nuclear repulsion energy | 47.417373 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3366 |
3249 |
35.00 |
|
|
|
| 2 |
A' |
1784 |
1722 |
39.06 |
|
|
|
| 3 |
A' |
1215 |
1173 |
7.94 |
|
|
|
| 4 |
A' |
479 |
462 |
23.60 |
|
|
|
| 5 |
A' |
389 |
376 |
18.49 |
|
|
|
| 6 |
A" |
437 |
422 |
0.34 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3835.0 cm
-1
Scaled (by 0.9652) Zero Point Vibrational Energy (zpe) 3701.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/aug-cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.112 |
-1.215 |
0.000 |
| C2 |
0.000 |
0.087 |
0.000 |
| N3 |
-0.195 |
1.269 |
0.000 |
| H4 |
0.689 |
-2.116 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3073 | 2.5028 | 1.0695 |
C2 | 1.3073 | | 1.1974 | 2.3084 | N3 | 2.5028 | 1.1974 | | 3.4981 | H4 | 1.0695 | 2.3084 | 3.4981 | |
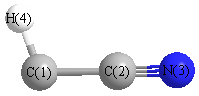 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
175.569 |
|
C2 |
C1 |
H4 |
152.297 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S2C1
S2C2
Energy calculated at TPSSh/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -131.477665 |
| Energy at 298.15K | |
| HF Energy | -131.477665 |
| Nuclear repulsion energy | 47.482793 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3428 |
3309 |
65.93 |
|
|
|
| 2 |
Σ |
1720 |
1660 |
40.55 |
|
|
|
| 3 |
Σ |
1255 |
1212 |
4.99 |
|
|
|
| 4 |
Π |
436 |
421 |
0.00 |
|
|
|
| 4 |
Π |
436 |
421 |
0.00 |
|
|
|
| 5 |
Π |
303i |
293i |
44.39 |
|
|
|
| 5 |
Π |
303i |
293i |
44.39 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3334.7 cm
-1
Scaled (by 0.9652) Zero Point Vibrational Energy (zpe) 3218.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/aug-cc-pVTZ
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.207 |
| C2 |
0.000 |
0.000 |
0.083 |
| N3 |
0.000 |
0.000 |
1.288 |
| H4 |
0.000 |
0.000 |
-2.272 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.2895 | 2.4953 | 1.0647 |
C2 | 1.2895 | | 1.2059 | 2.3542 | N3 | 2.4953 | 1.2059 | | 3.5601 | H4 | 1.0647 | 2.3542 | 3.5601 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
180.000 |
|
C2 |
C1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.565 |
|
|
|
| 2 |
C |
0.825 |
|
|
|
| 3 |
N |
-0.602 |
|
|
|
| 4 |
H |
0.342 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.305 |
3.305 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.106 |
0.000 |
0.000 |
| y |
0.000 |
3.106 |
0.000 |
| z |
0.000 |
0.000 |
7.117 |
<r2> (average value of r
2) Å
2
| <r2> |
36.044 |
| (<r2>)1/2 |
6.004 |
Jump to
S1C1
S1C2
S2C2
Energy calculated at TPSSh/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -131.448326 |
| Energy at 298.15K | -131.447766 |
| HF Energy | -131.448326 |
| Nuclear repulsion energy | 47.074228 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3057 |
2951 |
5.09 |
|
|
|
| 2 |
A' |
2063 |
1992 |
22.78 |
|
|
|
| 3 |
A' |
1087 |
1049 |
42.68 |
|
|
|
| 4 |
A' |
945 |
912 |
41.46 |
|
|
|
| 5 |
A' |
457 |
441 |
23.11 |
|
|
|
| 6 |
A" |
303 |
293 |
9.55 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3956.1 cm
-1
Scaled (by 0.9652) Zero Point Vibrational Energy (zpe) 3818.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/aug-cc-pVTZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.149 |
-1.271 |
0.000 |
| C2 |
0.000 |
0.092 |
0.000 |
| N3 |
-0.300 |
1.233 |
0.000 |
| H4 |
1.207 |
-1.565 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3712 | 2.5439 | 1.0985 |
C2 | 1.3712 | | 1.1797 | 2.0505 | N3 | 2.5439 | 1.1797 | | 3.1785 | H4 | 1.0985 | 2.0505 | 3.1785 | |
More geometry information
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.395 |
|
|
|
| 2 |
C |
0.451 |
|
|
|
| 3 |
N |
-0.309 |
|
|
|
| 4 |
H |
0.253 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.770 |
-1.315 |
0.000 |
2.205 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.615 |
-0.728 |
0.000 |
| y |
-0.728 |
7.049 |
0.000 |
| z |
0.000 |
0.000 |
3.371 |
<r2> (average value of r
2) Å
2
| <r2> |
36.340 |
| (<r2>)1/2 |
6.028 |
Jump to
S1C1
S1C2
S2C1
Energy calculated at TPSSh/aug-cc-pVTZ
| | hartrees |
|---|
| Energy at 0K | -131.448326 |
| Energy at 298.15K | -131.447766 |
| HF Energy | -131.448326 |
| Nuclear repulsion energy | 47.074228 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/aug-cc-pVTZ
Geometric Data calculated at TPSSh/aug-cc-pVTZ
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability