Jump to
S1C2
Energy calculated at TPSSh/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -485.362917 |
| Energy at 298.15K | |
| HF Energy | -485.362917 |
| Nuclear repulsion energy | 315.918481 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
1753 |
1687 |
365.12 |
|
|
|
| 2 |
A' |
1389 |
1337 |
61.89 |
|
|
|
| 3 |
A' |
1286 |
1238 |
360.07 |
|
|
|
| 4 |
A' |
870 |
838 |
56.66 |
|
|
|
| 5 |
A' |
788 |
758 |
7.12 |
|
|
|
| 6 |
A' |
726 |
699 |
277.78 |
|
|
|
| 7 |
A' |
618 |
595 |
76.74 |
|
|
|
| 8 |
A' |
499 |
480 |
236.14 |
|
|
|
| 9 |
A' |
308 |
296 |
178.02 |
|
|
|
| 10 |
A' |
175 |
168 |
0.96 |
|
|
|
| 11 |
A" |
1848 |
1779 |
400.86 |
|
|
|
| 12 |
A" |
724 |
697 |
8.31 |
|
|
|
| 13 |
A" |
492 |
474 |
2.13 |
|
|
|
| 14 |
A" |
84 |
81 |
0.05 |
|
|
|
| 15 |
A" |
24i |
23i |
0.02 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5768.1 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 5551.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.896 |
-0.159 |
0.000 |
| N2 |
-0.007 |
-1.216 |
0.000 |
| N3 |
0.247 |
1.081 |
0.000 |
| O4 |
0.514 |
-2.260 |
0.000 |
| O5 |
-0.007 |
1.665 |
1.436 |
| O6 |
-1.606 |
-0.792 |
0.000 |
| O7 |
-0.007 |
1.665 |
-1.436 |
Atom - Atom Distances (Å)
| |
O1 |
N2 |
N3 |
O4 |
O5 |
O6 |
O7 |
| O1 | | 1.3906 | 1.3995 | 2.1360 | 2.4910 | 2.5812 | 2.4910 |
N2 | 1.3906 | | 2.3113 | 1.1665 | 3.2197 | 1.6547 | 3.2197 | N3 | 1.3995 | 2.3113 | | 3.3520 | 1.5712 | 2.6349 | 1.5712 | O4 | 2.1360 | 1.1665 | 3.3520 | | 4.2123 | 2.5787 | 4.2123 | O5 | 2.4910 | 3.2197 | 1.5712 | 4.2123 | | 3.2651 | 2.8726 | O6 | 2.5812 | 1.6547 | 2.6349 | 2.5787 | 3.2651 | | 3.2651 | O7 | 2.4910 | 3.2197 | 1.5712 | 4.2123 | 2.8726 | 3.2651 | |
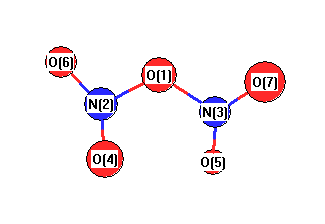 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
N2 |
O4 |
113.009 |
|
O1 |
N2 |
O6 |
115.635 |
| O1 |
N3 |
O5 |
113.846 |
|
O1 |
N3 |
O7 |
113.846 |
| N2 |
O1 |
N3 |
111.875 |
|
O4 |
N2 |
O6 |
131.356 |
| O5 |
N3 |
O7 |
132.173 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
0.151 |
|
|
|
| 2 |
N |
-0.079 |
|
|
|
| 3 |
N |
-0.140 |
|
|
|
| 4 |
O |
-0.022 |
|
|
|
| 5 |
O |
0.061 |
|
|
|
| 6 |
O |
-0.031 |
|
|
|
| 7 |
O |
0.061 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.250 |
-0.972 |
0.000 |
1.004 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.337 |
-0.710 |
0.000 |
| y |
-0.710 |
9.011 |
0.000 |
| z |
0.000 |
0.000 |
5.427 |
<r2> (average value of r
2) Å
2
| <r2> |
179.569 |
| (<r2>)1/2 |
13.400 |
Jump to
S1C1
Energy calculated at TPSSh/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -485.363728 |
| Energy at 298.15K | -485.367531 |
| HF Energy | -485.363728 |
| Nuclear repulsion energy | 314.208959 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
1827 |
1758 |
435.34 |
|
|
|
| 2 |
A |
1385 |
1333 |
39.29 |
|
|
|
| 3 |
A |
861 |
829 |
27.15 |
|
|
|
| 4 |
A |
781 |
751 |
0.86 |
|
|
|
| 5 |
A |
664 |
639 |
0.00 |
|
|
|
| 6 |
A |
364 |
351 |
1.42 |
|
|
|
| 7 |
A |
213 |
205 |
0.30 |
|
|
|
| 8 |
A |
57 |
55 |
0.29 |
|
|
|
| 9 |
B |
1782 |
1715 |
311.22 |
|
|
|
| 10 |
B |
1291 |
1243 |
377.22 |
|
|
|
| 11 |
B |
726 |
699 |
275.46 |
|
|
|
| 12 |
B |
710 |
683 |
17.44 |
|
|
|
| 13 |
B |
523 |
503 |
234.84 |
|
|
|
| 14 |
B |
297 |
286 |
375.35 |
|
|
|
| 15 |
B |
58 |
56 |
4.61 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5769.4 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 5553.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/6-31+G**
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.798 |
| N2 |
0.000 |
1.233 |
0.021 |
| N3 |
0.000 |
-1.233 |
0.021 |
| O4 |
0.616 |
2.101 |
0.570 |
| O5 |
-0.616 |
-2.101 |
0.570 |
| O6 |
-0.650 |
1.220 |
-0.988 |
| O7 |
0.650 |
-1.220 |
-0.988 |
Atom - Atom Distances (Å)
| |
O1 |
N2 |
N3 |
O4 |
O5 |
O6 |
O7 |
| O1 | | 1.4576 | 1.4576 | 2.2010 | 2.2010 | 2.2583 | 2.2583 |
N2 | 1.4576 | | 2.4667 | 1.1970 | 3.4345 | 1.2001 | 2.7312 | N3 | 1.4576 | 2.4667 | | 3.4345 | 1.1970 | 2.7312 | 1.2001 | O4 | 2.2010 | 1.1970 | 3.4345 | | 4.3783 | 2.1916 | 3.6680 | O5 | 2.2010 | 3.4345 | 1.1970 | 4.3783 | | 3.6680 | 2.1916 | O6 | 2.2583 | 1.2001 | 2.7312 | 2.1916 | 3.6680 | | 2.7648 | O7 | 2.2583 | 2.7312 | 1.2001 | 3.6680 | 2.1916 | 2.7648 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
N2 |
O4 |
111.648 |
|
O1 |
N2 |
O6 |
116.024 |
| O1 |
N3 |
O5 |
111.648 |
|
O1 |
N3 |
O7 |
116.024 |
| N2 |
O1 |
N3 |
115.588 |
|
O4 |
N2 |
O6 |
132.213 |
| O5 |
N3 |
O7 |
132.213 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
0.073 |
|
|
|
| 2 |
N |
-0.025 |
|
|
|
| 3 |
N |
-0.025 |
|
|
|
| 4 |
O |
-0.007 |
|
|
|
| 5 |
O |
-0.007 |
|
|
|
| 6 |
O |
-0.004 |
|
|
|
| 7 |
O |
-0.004 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.053 |
0.053 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.172 |
0.892 |
0.000 |
| y |
0.892 |
9.343 |
0.000 |
| z |
0.000 |
0.000 |
6.433 |
<r2> (average value of r
2) Å
2
| <r2> |
185.755 |
| (<r2>)1/2 |
13.629 |