Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3724 |
3584 |
67.41 |
|
|
|
| 2 |
A' |
3229 |
3108 |
3.61 |
|
|
|
| 3 |
A' |
3220 |
3099 |
18.12 |
|
|
|
| 4 |
A' |
3196 |
3076 |
10.59 |
|
|
|
| 5 |
A' |
3181 |
3062 |
14.98 |
|
|
|
| 6 |
A' |
1651 |
1589 |
84.81 |
|
|
|
| 7 |
A' |
1628 |
1567 |
92.37 |
|
|
|
| 8 |
A' |
1511 |
1455 |
117.18 |
|
|
|
| 9 |
A' |
1488 |
1433 |
56.49 |
|
|
|
| 10 |
A' |
1372 |
1321 |
32.88 |
|
|
|
| 11 |
A' |
1336 |
1286 |
21.36 |
|
|
|
| 12 |
A' |
1322 |
1272 |
60.74 |
|
|
|
| 13 |
A' |
1195 |
1150 |
138.07 |
|
|
|
| 14 |
A' |
1169 |
1125 |
35.31 |
|
|
|
| 15 |
A' |
1106 |
1065 |
29.00 |
|
|
|
| 16 |
A' |
1062 |
1022 |
3.48 |
|
|
|
| 17 |
A' |
1000 |
963 |
6.45 |
|
|
|
| 18 |
A' |
856 |
824 |
13.74 |
|
|
|
| 19 |
A' |
628 |
605 |
2.38 |
|
|
|
| 20 |
A' |
557 |
536 |
0.42 |
|
|
|
| 21 |
A' |
409 |
394 |
17.71 |
|
|
|
| 22 |
A" |
997 |
959 |
0.00 |
|
|
|
| 23 |
A" |
970 |
933 |
0.61 |
|
|
|
| 24 |
A" |
876 |
843 |
1.01 |
|
|
|
| 25 |
A" |
786 |
756 |
67.67 |
|
|
|
| 26 |
A" |
744 |
716 |
1.98 |
|
|
|
| 27 |
A" |
557 |
536 |
40.66 |
|
|
|
| 28 |
A" |
496 |
477 |
87.34 |
|
|
|
| 29 |
A" |
415 |
400 |
0.50 |
|
|
|
| 30 |
A" |
211 |
203 |
0.98 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20445.4 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 19678.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
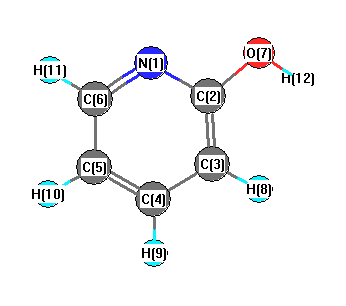
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.249 |
|
|
|
| 2 |
C |
0.017 |
|
|
|
| 3 |
C |
0.286 |
|
|
|
| 4 |
C |
-0.358 |
|
|
|
| 5 |
C |
0.039 |
|
|
|
| 6 |
C |
-0.274 |
|
|
|
| 7 |
O |
-0.460 |
|
|
|
| 8 |
H |
0.167 |
|
|
|
| 9 |
H |
0.160 |
|
|
|
| 10 |
H |
0.156 |
|
|
|
| 11 |
H |
0.148 |
|
|
|
| 12 |
H |
0.368 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.505 |
-1.345 |
0.000 |
1.436 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.121 |
-0.020 |
0.000 |
| y |
-0.020 |
12.895 |
0.000 |
| z |
0.000 |
0.000 |
5.734 |
<r2> (average value of r
2) Å
2
| <r2> |
177.577 |
| (<r2>)1/2 |
13.326 |