Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3672 |
3534 |
60.90 |
|
|
|
| 2 |
A' |
3278 |
3155 |
4.92 |
|
|
|
| 3 |
A' |
3257 |
3135 |
2.22 |
|
|
|
| 4 |
A' |
3208 |
3088 |
24.08 |
|
|
|
| 5 |
A' |
3196 |
3076 |
40.55 |
|
|
|
| 6 |
A' |
3185 |
3066 |
5.02 |
|
|
|
| 7 |
A' |
3178 |
3059 |
1.87 |
|
|
|
| 8 |
A' |
1661 |
1598 |
2.81 |
|
|
|
| 9 |
A' |
1621 |
1560 |
0.84 |
|
|
|
| 10 |
A' |
1553 |
1495 |
8.64 |
|
|
|
| 11 |
A' |
1526 |
1469 |
2.62 |
|
|
|
| 12 |
A' |
1488 |
1432 |
21.54 |
|
|
|
| 13 |
A' |
1455 |
1400 |
12.75 |
|
|
|
| 14 |
A' |
1390 |
1337 |
25.09 |
|
|
|
| 15 |
A' |
1382 |
1330 |
12.79 |
|
|
|
| 16 |
A' |
1302 |
1253 |
14.66 |
|
|
|
| 17 |
A' |
1270 |
1222 |
10.88 |
|
|
|
| 18 |
A' |
1225 |
1179 |
3.43 |
|
|
|
| 19 |
A' |
1176 |
1132 |
1.25 |
|
|
|
| 20 |
A' |
1145 |
1102 |
0.70 |
|
|
|
| 21 |
A' |
1112 |
1070 |
25.60 |
|
|
|
| 22 |
A' |
1091 |
1050 |
7.37 |
|
|
|
| 23 |
A' |
1033 |
995 |
7.11 |
|
|
|
| 24 |
A' |
901 |
867 |
6.18 |
|
|
|
| 25 |
A' |
879 |
846 |
0.44 |
|
|
|
| 26 |
A' |
768 |
739 |
2.49 |
|
|
|
| 27 |
A' |
607 |
584 |
1.34 |
|
|
|
| 28 |
A' |
545 |
524 |
0.06 |
|
|
|
| 29 |
A' |
398 |
383 |
4.45 |
|
|
|
| 30 |
A" |
974 |
937 |
0.03 |
|
|
|
| 31 |
A" |
935 |
900 |
1.98 |
|
|
|
| 32 |
A" |
857 |
825 |
3.69 |
|
|
|
| 33 |
A" |
851 |
819 |
0.28 |
|
|
|
| 34 |
A" |
776 |
747 |
16.84 |
|
|
|
| 35 |
A" |
750 |
722 |
101.92 |
|
|
|
| 36 |
A" |
726 |
698 |
37.54 |
|
|
|
| 37 |
A" |
609 |
586 |
8.23 |
|
|
|
| 38 |
A" |
579 |
557 |
1.03 |
|
|
|
| 39 |
A" |
427 |
411 |
3.64 |
|
|
|
| 40 |
A" |
393 |
378 |
69.56 |
|
|
|
| 41 |
A" |
242 |
233 |
0.05 |
|
|
|
| 42 |
A" |
213 |
205 |
12.33 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 28414.8 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 27349.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
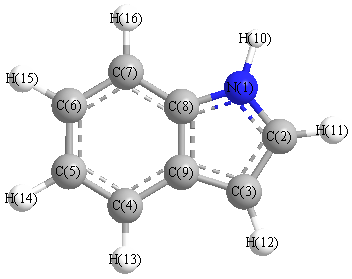
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.248 |
|
|
|
| 2 |
C |
-0.121 |
|
|
|
| 3 |
C |
0.096 |
|
|
|
| 4 |
C |
-0.665 |
|
|
|
| 5 |
C |
0.077 |
|
|
|
| 6 |
C |
-0.452 |
|
|
|
| 7 |
C |
0.188 |
|
|
|
| 8 |
C |
-1.568 |
|
|
|
| 9 |
C |
1.501 |
|
|
|
| 10 |
H |
0.314 |
|
|
|
| 11 |
H |
0.153 |
|
|
|
| 12 |
H |
0.152 |
|
|
|
| 13 |
H |
0.148 |
|
|
|
| 14 |
H |
0.142 |
|
|
|
| 15 |
H |
0.144 |
|
|
|
| 16 |
H |
0.138 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.894 |
1.992 |
0.000 |
2.183 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
19.855 |
-1.598 |
0.000 |
| y |
-1.598 |
15.913 |
0.000 |
| z |
0.000 |
0.000 |
8.170 |
<r2> (average value of r
2) Å
2
| <r2> |
283.662 |
| (<r2>)1/2 |
16.842 |