Jump to
S1C2
Vibrational Frequencies calculated at TPSSh/6-31+G**
Geometric Data calculated at TPSSh/6-31+G**
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at TPSSh/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -235.886733 |
| Energy at 298.15K | -235.900101 |
| HF Energy | -235.886733 |
| Nuclear repulsion energy | 254.343883 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3121 |
3004 |
83.70 |
|
|
|
| 2 |
A' |
3100 |
2983 |
54.19 |
|
|
|
| 3 |
A' |
3097 |
2981 |
34.77 |
|
|
|
| 4 |
A' |
3090 |
2974 |
16.54 |
|
|
|
| 5 |
A' |
3067 |
2952 |
43.90 |
|
|
|
| 6 |
A' |
3047 |
2932 |
12.59 |
|
|
|
| 7 |
A' |
3025 |
2912 |
43.63 |
|
|
|
| 8 |
A' |
3020 |
2907 |
44.65 |
|
|
|
| 9 |
A' |
1527 |
1470 |
2.04 |
|
|
|
| 10 |
A' |
1520 |
1463 |
1.76 |
|
|
|
| 11 |
A' |
1510 |
1453 |
6.04 |
|
|
|
| 12 |
A' |
1496 |
1439 |
0.11 |
|
|
|
| 13 |
A' |
1427 |
1374 |
1.04 |
|
|
|
| 14 |
A' |
1410 |
1357 |
7.47 |
|
|
|
| 15 |
A' |
1317 |
1268 |
5.83 |
|
|
|
| 16 |
A' |
1302 |
1253 |
3.63 |
|
|
|
| 17 |
A' |
1232 |
1186 |
1.94 |
|
|
|
| 18 |
A' |
1208 |
1163 |
0.54 |
|
|
|
| 19 |
A' |
1009 |
971 |
0.17 |
|
|
|
| 20 |
A' |
986 |
949 |
0.61 |
|
|
|
| 21 |
A' |
948 |
913 |
0.19 |
|
|
|
| 22 |
A' |
920 |
886 |
0.19 |
|
|
|
| 23 |
A' |
871 |
838 |
0.19 |
|
|
|
| 24 |
A' |
704 |
678 |
0.93 |
|
|
|
| 25 |
A' |
595 |
573 |
1.18 |
|
|
|
| 26 |
A' |
384 |
369 |
0.62 |
|
|
|
| 27 |
A' |
338 |
325 |
0.02 |
|
|
|
| 28 |
A' |
161 |
155 |
0.00 |
|
|
|
| 29 |
A" |
3097 |
2981 |
44.65 |
|
|
|
| 30 |
A" |
3094 |
2978 |
36.17 |
|
|
|
| 31 |
A" |
3089 |
2974 |
9.82 |
|
|
|
| 32 |
A" |
3042 |
2928 |
103.36 |
|
|
|
| 33 |
A" |
1525 |
1467 |
7.58 |
|
|
|
| 34 |
A" |
1502 |
1445 |
0.09 |
|
|
|
| 35 |
A" |
1491 |
1435 |
2.39 |
|
|
|
| 36 |
A" |
1280 |
1232 |
0.32 |
|
|
|
| 37 |
A" |
1258 |
1211 |
0.13 |
|
|
|
| 38 |
A" |
1226 |
1180 |
0.02 |
|
|
|
| 39 |
A" |
1185 |
1140 |
1.60 |
|
|
|
| 40 |
A" |
1088 |
1048 |
0.04 |
|
|
|
| 41 |
A" |
984 |
947 |
0.00 |
|
|
|
| 42 |
A" |
947 |
911 |
2.06 |
|
|
|
| 43 |
A" |
878 |
845 |
0.00 |
|
|
|
| 44 |
A" |
785 |
756 |
0.16 |
|
|
|
| 45 |
A" |
377 |
362 |
0.00 |
|
|
|
| 46 |
A" |
292 |
281 |
0.01 |
|
|
|
| 47 |
A" |
261 |
251 |
0.00 |
|
|
|
| 48 |
A" |
223 |
214 |
0.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36525.7 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 35156.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.095 |
0.404 |
0.000 |
| C2 |
0.353 |
-1.737 |
0.000 |
| C3 |
-0.095 |
-0.721 |
1.084 |
| C4 |
-0.095 |
-0.721 |
-1.084 |
| C5 |
1.218 |
1.198 |
0.000 |
| C6 |
-1.297 |
1.349 |
0.000 |
| H7 |
1.436 |
-1.899 |
0.000 |
| H8 |
-0.141 |
-2.713 |
0.000 |
| H9 |
0.567 |
-0.576 |
1.946 |
| H10 |
0.567 |
-0.576 |
-1.946 |
| H11 |
-1.105 |
-0.931 |
1.454 |
| H12 |
-1.105 |
-0.931 |
-1.454 |
| H13 |
2.089 |
0.532 |
0.000 |
| H14 |
-2.239 |
0.788 |
0.000 |
| H15 |
1.285 |
1.838 |
-0.888 |
| H16 |
1.285 |
1.838 |
0.888 |
| H17 |
-1.289 |
1.996 |
0.887 |
| H18 |
-1.289 |
1.996 |
-0.887 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
C5 |
C6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
| C1 | | 2.1873 | 1.5627 | 1.5627 | 1.5339 | 1.5295 | 2.7653 | 3.1174 | 2.2773 | 2.2773 | 2.2173 | 2.2173 | 2.1875 | 2.1785 | 2.1792 | 2.1792 | 2.1784 | 2.1784 |
C2 | 2.1873 | | 1.5518 | 1.5518 | 3.0589 | 3.4995 | 1.0945 | 1.0941 | 2.2754 | 2.2754 | 2.2108 | 2.2108 | 2.8561 | 3.6187 | 3.7995 | 3.7995 | 4.1729 | 4.1729 | C3 | 1.5627 | 1.5518 | | 2.1687 | 2.5651 | 2.6282 | 2.2148 | 2.2683 | 1.0960 | 3.1049 | 1.0958 | 2.7396 | 2.7411 | 2.8375 | 3.5133 | 2.9142 | 2.9741 | 3.5625 | C4 | 1.5627 | 1.5518 | 2.1687 | | 2.5651 | 2.6282 | 2.2148 | 2.2683 | 3.1049 | 1.0960 | 2.7396 | 1.0958 | 2.7411 | 2.8375 | 2.9142 | 3.5133 | 3.5625 | 2.9741 | C5 | 1.5339 | 3.0589 | 2.5651 | 2.5651 | | 2.5201 | 3.1041 | 4.1397 | 2.7122 | 2.7122 | 3.4699 | 3.4699 | 1.0965 | 3.4814 | 1.0969 | 1.0969 | 2.7767 | 2.7767 | C6 | 1.5295 | 3.4995 | 2.6282 | 2.6282 | 2.5201 | | 4.2449 | 4.2234 | 3.3123 | 3.3123 | 2.7110 | 2.7110 | 3.4839 | 1.0962 | 2.7745 | 2.7745 | 1.0973 | 1.0973 | H7 | 2.7653 | 1.0945 | 2.2148 | 2.2148 | 3.1041 | 4.2449 | | 1.7741 | 2.5077 | 2.5077 | 3.0828 | 3.0828 | 2.5167 | 4.5524 | 3.8441 | 3.8441 | 4.8351 | 4.8351 | H8 | 3.1174 | 1.0941 | 2.2683 | 2.2683 | 4.1397 | 4.2234 | 1.7741 | | 2.9751 | 2.9751 | 2.4933 | 2.4933 | 3.9367 | 4.0816 | 4.8511 | 4.8511 | 4.9268 | 4.9268 | H9 | 2.2773 | 2.2754 | 1.0960 | 3.1049 | 2.7122 | 3.3123 | 2.5077 | 2.9751 | | 3.8914 | 1.7789 | 3.8049 | 2.7072 | 3.6776 | 3.7914 | 2.7323 | 3.3443 | 4.2527 | H10 | 2.2773 | 2.2754 | 3.1049 | 1.0960 | 2.7122 | 3.3123 | 2.5077 | 2.9751 | 3.8914 | | 3.8049 | 1.7789 | 2.7072 | 3.6776 | 2.7323 | 3.7914 | 4.2527 | 3.3443 | H11 | 2.2173 | 2.2108 | 1.0958 | 2.7396 | 3.4699 | 2.7110 | 3.0828 | 2.4933 | 1.7789 | 3.8049 | | 2.9071 | 3.8018 | 2.5210 | 4.3433 | 3.7016 | 2.9868 | 3.7518 | H12 | 2.2173 | 2.2108 | 2.7396 | 1.0958 | 3.4699 | 2.7110 | 3.0828 | 2.4933 | 3.8049 | 1.7789 | 2.9071 | | 3.8018 | 2.5210 | 3.7016 | 4.3433 | 3.7518 | 2.9868 | H13 | 2.1875 | 2.8561 | 2.7411 | 2.7411 | 1.0965 | 3.4839 | 2.5167 | 3.9367 | 2.7072 | 2.7072 | 3.8018 | 3.8018 | | 4.3359 | 1.7725 | 1.7725 | 3.7873 | 3.7873 | H14 | 2.1785 | 3.6187 | 2.8375 | 2.8375 | 3.4814 | 1.0962 | 4.5524 | 4.0816 | 3.6776 | 3.6776 | 2.5210 | 2.5210 | 4.3359 | | 3.7832 | 3.7832 | 1.7738 | 1.7738 | H15 | 2.1792 | 3.7995 | 3.5133 | 2.9142 | 1.0969 | 2.7745 | 3.8441 | 4.8511 | 3.7914 | 2.7323 | 4.3433 | 3.7016 | 1.7725 | 3.7832 | | 1.7756 | 3.1308 | 2.5794 | H16 | 2.1792 | 3.7995 | 2.9142 | 3.5133 | 1.0969 | 2.7745 | 3.8441 | 4.8511 | 2.7323 | 3.7914 | 3.7016 | 4.3433 | 1.7725 | 3.7832 | 1.7756 | | 2.5794 | 3.1308 | H17 | 2.1784 | 4.1729 | 2.9741 | 3.5625 | 2.7767 | 1.0973 | 4.8351 | 4.9268 | 3.3443 | 4.2527 | 2.9868 | 3.7518 | 3.7873 | 1.7738 | 3.1308 | 2.5794 | | 1.7733 | H18 | 2.1784 | 4.1729 | 3.5625 | 2.9741 | 2.7767 | 1.0973 | 4.8351 | 4.9268 | 4.2527 | 3.3443 | 3.7518 | 2.9868 | 3.7873 | 1.7738 | 2.5794 | 3.1308 | 1.7733 | |
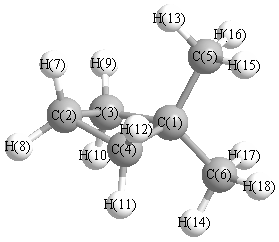 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C3 |
C2 |
89.222 |
|
C1 |
C3 |
H9 |
116.769 |
| C1 |
C3 |
H11 |
111.834 |
|
C1 |
C4 |
C2 |
89.222 |
| C1 |
C4 |
H10 |
116.769 |
|
C1 |
C4 |
H12 |
111.834 |
| C1 |
C5 |
H13 |
111.456 |
|
C1 |
C5 |
H15 |
110.762 |
| C1 |
C5 |
H16 |
110.762 |
|
C1 |
C6 |
H14 |
111.064 |
| C1 |
C6 |
H17 |
110.984 |
|
C1 |
C6 |
H18 |
110.984 |
| C2 |
C3 |
H9 |
117.452 |
|
C2 |
C3 |
H11 |
112.089 |
| C2 |
C4 |
H10 |
117.452 |
|
C2 |
C4 |
H12 |
112.089 |
| C3 |
C1 |
C4 |
87.879 |
|
C3 |
C1 |
C5 |
111.863 |
| C3 |
C1 |
C6 |
116.412 |
|
C3 |
C2 |
C4 |
88.655 |
| C3 |
C2 |
H7 |
112.494 |
|
C3 |
C2 |
H8 |
116.975 |
| C4 |
C1 |
C5 |
111.863 |
|
C4 |
C1 |
C6 |
116.412 |
| C4 |
C2 |
H7 |
112.494 |
|
C4 |
C2 |
H8 |
116.975 |
| C5 |
C1 |
C6 |
110.706 |
|
H7 |
C2 |
H8 |
108.316 |
| H9 |
C3 |
H11 |
108.504 |
|
H10 |
C4 |
H12 |
108.504 |
| H13 |
C5 |
H15 |
107.828 |
|
H13 |
C5 |
H16 |
107.828 |
| H14 |
C6 |
H17 |
107.930 |
|
H14 |
C6 |
H18 |
107.930 |
| H15 |
C5 |
H16 |
108.067 |
|
H17 |
C6 |
H18 |
107.807 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.663 |
|
|
|
| 2 |
C |
-0.235 |
|
|
|
| 3 |
C |
-0.479 |
|
|
|
| 4 |
C |
-0.479 |
|
|
|
| 5 |
C |
-0.677 |
|
|
|
| 6 |
C |
-0.563 |
|
|
|
| 7 |
H |
0.152 |
|
|
|
| 8 |
H |
0.145 |
|
|
|
| 9 |
H |
0.145 |
|
|
|
| 10 |
H |
0.145 |
|
|
|
| 11 |
H |
0.146 |
|
|
|
| 12 |
H |
0.146 |
|
|
|
| 13 |
H |
0.149 |
|
|
|
| 14 |
H |
0.145 |
|
|
|
| 15 |
H |
0.151 |
|
|
|
| 16 |
H |
0.151 |
|
|
|
| 17 |
H |
0.148 |
|
|
|
| 18 |
H |
0.148 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.013 |
0.012 |
0.000 |
0.018 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.054 |
-0.198 |
0.000 |
| y |
-0.198 |
11.071 |
0.000 |
| z |
0.000 |
0.000 |
10.046 |
<r2> (average value of r
2) Å
2
| <r2> |
169.142 |
| (<r2>)1/2 |
13.005 |