Jump to
S1C2
S2C1
S2C2
Energy calculated at TPSSh/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -131.438171 |
| Energy at 298.15K | -131.437494 |
| HF Energy | -131.438171 |
| Nuclear repulsion energy | 47.093509 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3391 |
3264 |
33.26 |
|
|
|
| 2 |
A' |
1810 |
1742 |
33.99 |
|
|
|
| 3 |
A' |
1218 |
1172 |
7.79 |
|
|
|
| 4 |
A' |
498 |
479 |
33.78 |
|
|
|
| 5 |
A' |
393 |
378 |
13.40 |
|
|
|
| 6 |
A" |
434 |
417 |
0.44 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3871.3 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 3726.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.109 |
-1.225 |
0.000 |
| C2 |
0.000 |
0.087 |
0.000 |
| N3 |
-0.195 |
1.278 |
0.000 |
| H4 |
0.712 |
-2.112 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3171 | 2.5213 | 1.0725 |
C2 | 1.3171 | | 1.2063 | 2.3118 | N3 | 2.5213 | 1.2063 | | 3.5091 | H4 | 1.0725 | 2.3118 | 3.5091 | |
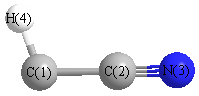 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
175.437 |
|
C2 |
C1 |
H4 |
150.526 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
S2C1
S2C2
Energy calculated at TPSSh/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -131.437335 |
| Energy at 298.15K | |
| HF Energy | -131.437335 |
| Nuclear repulsion energy | 47.159096 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Σ |
3453 |
3323 |
68.67 |
|
|
|
| 2 |
Σ |
1742 |
1677 |
36.12 |
|
|
|
| 3 |
Σ |
1262 |
1214 |
4.90 |
|
|
|
| 4 |
Π |
442 |
425 |
0.10 |
|
|
|
| 4 |
Π |
442 |
425 |
0.10 |
|
|
|
| 5 |
Π |
306i |
295i |
60.55 |
|
|
|
| 5 |
Π |
306i |
295i |
60.55 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3363.8 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 3237.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/6-31+G**
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.216 |
| C2 |
0.000 |
0.000 |
0.082 |
| N3 |
0.000 |
0.000 |
1.298 |
| H4 |
0.000 |
0.000 |
-2.283 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.2977 | 2.5132 | 1.0677 |
C2 | 1.2977 | | 1.2155 | 2.3655 | N3 | 2.5132 | 1.2155 | | 3.5809 | H4 | 1.0677 | 2.3655 | 3.5809 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
N3 |
180.000 |
|
C2 |
C1 |
H4 |
180.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.330 |
|
|
|
| 2 |
C |
-0.154 |
|
|
|
| 3 |
N |
-0.440 |
|
|
|
| 4 |
H |
0.265 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-3.366 |
3.366 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.202 |
0.000 |
0.000 |
| y |
0.000 |
2.202 |
0.000 |
| z |
0.000 |
0.000 |
7.065 |
<r2> (average value of r
2) Å
2
| <r2> |
36.426 |
| (<r2>)1/2 |
6.035 |
Jump to
S1C1
S1C2
S2C2
Energy calculated at TPSSh/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -131.407330 |
| Energy at 298.15K | -131.406736 |
| HF Energy | -131.407330 |
| Nuclear repulsion energy | 46.768975 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3070 |
2955 |
9.69 |
|
|
|
| 2 |
A' |
2074 |
1996 |
18.41 |
|
|
|
| 3 |
A' |
1086 |
1046 |
38.13 |
|
|
|
| 4 |
A' |
931 |
896 |
48.77 |
|
|
|
| 5 |
A' |
433 |
417 |
25.72 |
|
|
|
| 6 |
A" |
302 |
291 |
6.21 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3947.9 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 3799.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at TPSSh/6-31+G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.246 |
-1.264 |
0.000 |
| C2 |
0.000 |
0.093 |
0.000 |
| N3 |
-0.401 |
1.213 |
0.000 |
| H4 |
1.327 |
-1.472 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
N3 |
H4 |
| C1 | | 1.3789 | 2.5601 | 1.1009 |
C2 | 1.3789 | | 1.1897 | 2.0522 | N3 | 2.5601 | 1.1897 | | 3.1934 | H4 | 1.1009 | 2.0522 | 3.1934 | |
More geometry information
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.109 |
|
|
|
| 2 |
C |
0.246 |
|
|
|
| 3 |
N |
-0.310 |
|
|
|
| 4 |
H |
0.173 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.026 |
-1.138 |
0.000 |
2.324 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.183 |
-1.142 |
0.000 |
| y |
-1.142 |
6.927 |
0.000 |
| z |
0.000 |
0.000 |
2.717 |
<r2> (average value of r
2) Å
2
| <r2> |
36.719 |
| (<r2>)1/2 |
6.060 |
Jump to
S1C1
S1C2
S2C1
Energy calculated at TPSSh/6-31+G**
| | hartrees |
|---|
| Energy at 0K | -131.407330 |
| Energy at 298.15K | -131.406736 |
| HF Energy | -131.407330 |
| Nuclear repulsion energy | 46.768975 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at TPSSh/6-31+G**
Geometric Data calculated at TPSSh/6-31+G**
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability