Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3118 |
3001 |
29.87 |
|
|
|
| 2 |
A' |
3113 |
2996 |
43.21 |
|
|
|
| 3 |
A' |
3046 |
2932 |
32.01 |
|
|
|
| 4 |
A' |
3046 |
2931 |
34.36 |
|
|
|
| 5 |
A' |
3042 |
2928 |
37.40 |
|
|
|
| 6 |
A' |
3036 |
2922 |
48.87 |
|
|
|
| 7 |
A' |
3031 |
2917 |
15.22 |
|
|
|
| 8 |
A' |
1525 |
1467 |
3.85 |
|
|
|
| 9 |
A' |
1524 |
1466 |
2.77 |
|
|
|
| 10 |
A' |
1511 |
1454 |
0.86 |
|
|
|
| 11 |
A' |
1504 |
1448 |
1.41 |
|
|
|
| 12 |
A' |
1495 |
1439 |
9.10 |
|
|
|
| 13 |
A' |
1425 |
1371 |
1.15 |
|
|
|
| 14 |
A' |
1423 |
1370 |
2.88 |
|
|
|
| 15 |
A' |
1376 |
1324 |
1.18 |
|
|
|
| 16 |
A' |
1310 |
1261 |
21.49 |
|
|
|
| 17 |
A' |
1258 |
1211 |
40.91 |
|
|
|
| 18 |
A' |
1120 |
1078 |
2.70 |
|
|
|
| 19 |
A' |
1067 |
1027 |
4.40 |
|
|
|
| 20 |
A' |
1046 |
1007 |
0.19 |
|
|
|
| 21 |
A' |
992 |
955 |
4.53 |
|
|
|
| 22 |
A' |
904 |
871 |
2.56 |
|
|
|
| 23 |
A' |
757 |
729 |
2.18 |
|
|
|
| 24 |
A' |
684 |
658 |
1.29 |
|
|
|
| 25 |
A' |
373 |
359 |
0.36 |
|
|
|
| 26 |
A' |
295 |
284 |
0.70 |
|
|
|
| 27 |
A' |
263 |
253 |
1.06 |
|
|
|
| 28 |
A' |
101 |
97 |
0.28 |
|
|
|
| 29 |
A" |
3131 |
3013 |
31.16 |
|
|
|
| 30 |
A" |
3111 |
2994 |
72.47 |
|
|
|
| 31 |
A" |
3091 |
2976 |
0.02 |
|
|
|
| 32 |
A" |
3087 |
2971 |
16.42 |
|
|
|
| 33 |
A" |
3071 |
2955 |
2.42 |
|
|
|
| 34 |
A" |
1520 |
1463 |
7.66 |
|
|
|
| 35 |
A" |
1513 |
1456 |
8.77 |
|
|
|
| 36 |
A" |
1326 |
1276 |
0.49 |
|
|
|
| 37 |
A" |
1269 |
1222 |
0.04 |
|
|
|
| 38 |
A" |
1244 |
1198 |
0.05 |
|
|
|
| 39 |
A" |
1068 |
1028 |
0.68 |
|
|
|
| 40 |
A" |
1041 |
1002 |
0.11 |
|
|
|
| 41 |
A" |
867 |
834 |
0.08 |
|
|
|
| 42 |
A" |
788 |
758 |
3.80 |
|
|
|
| 43 |
A" |
744 |
716 |
2.47 |
|
|
|
| 44 |
A" |
246 |
236 |
0.07 |
|
|
|
| 45 |
A" |
238 |
229 |
0.02 |
|
|
|
| 46 |
A" |
112 |
108 |
1.08 |
|
|
|
| 47 |
A" |
61 |
59 |
0.46 |
|
|
|
| 48 |
A" |
40 |
38 |
0.30 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 35473.3 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 34143.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
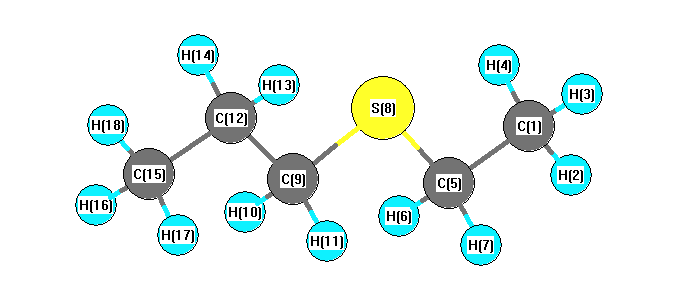
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.642 |
|
|
|
| 2 |
H |
0.161 |
|
|
|
| 3 |
H |
0.171 |
|
|
|
| 4 |
H |
0.171 |
|
|
|
| 5 |
C |
-0.159 |
|
|
|
| 6 |
H |
0.176 |
|
|
|
| 7 |
H |
0.176 |
|
|
|
| 8 |
S |
-0.148 |
|
|
|
| 9 |
C |
-0.235 |
|
|
|
| 10 |
H |
0.175 |
|
|
|
| 11 |
H |
0.175 |
|
|
|
| 12 |
C |
-0.252 |
|
|
|
| 13 |
H |
0.168 |
|
|
|
| 14 |
H |
0.168 |
|
|
|
| 15 |
C |
-0.574 |
|
|
|
| 16 |
H |
0.154 |
|
|
|
| 17 |
H |
0.154 |
|
|
|
| 18 |
H |
0.161 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.009 |
-1.372 |
0.000 |
1.703 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.265 |
2.373 |
0.000 |
| y |
2.373 |
13.117 |
0.000 |
| z |
0.000 |
0.000 |
9.860 |
<r2> (average value of r
2) Å
2
| <r2> |
356.881 |
| (<r2>)1/2 |
18.891 |