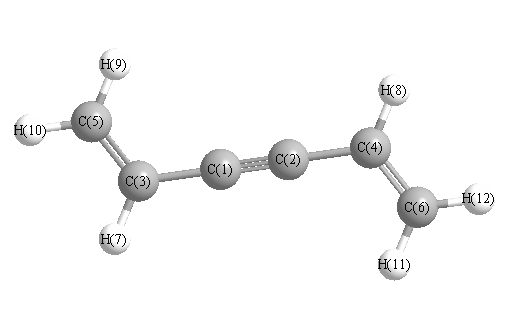
Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3265 |
3142 |
0.00 |
|
|
|
| 2 |
Ag |
3171 |
3052 |
0.00 |
|
|
|
| 3 |
Ag |
3154 |
3036 |
0.00 |
|
|
|
| 4 |
Ag |
2278 |
2192 |
0.00 |
|
|
|
| 5 |
Ag |
1644 |
1582 |
0.00 |
|
|
|
| 6 |
Ag |
1447 |
1393 |
0.00 |
|
|
|
| 7 |
Ag |
1325 |
1276 |
0.00 |
|
|
|
| 8 |
Ag |
1086 |
1045 |
0.00 |
|
|
|
| 9 |
Ag |
721 |
694 |
0.00 |
|
|
|
| 10 |
Ag |
522 |
502 |
0.00 |
|
|
|
| 11 |
Ag |
219 |
211 |
0.00 |
|
|
|
| 12 |
Au |
1005 |
967 |
48.08 |
|
|
|
| 13 |
Au |
936 |
901 |
91.37 |
|
|
|
| 14 |
Au |
681 |
656 |
14.12 |
|
|
|
| 15 |
Au |
183 |
176 |
7.54 |
|
|
|
| 16 |
Au |
52 |
50 |
0.17 |
|
|
|
| 17 |
Bg |
999 |
962 |
0.00 |
|
|
|
| 18 |
Bg |
937 |
902 |
0.00 |
|
|
|
| 19 |
Bg |
699 |
673 |
0.00 |
|
|
|
| 20 |
Bg |
372 |
358 |
0.00 |
|
|
|
| 21 |
Bu |
3265 |
3142 |
19.53 |
|
|
|
| 22 |
Bu |
3171 |
3052 |
12.11 |
|
|
|
| 23 |
Bu |
3154 |
3036 |
21.97 |
|
|
|
| 24 |
Bu |
1674 |
1611 |
43.02 |
|
|
|
| 25 |
Bu |
1471 |
1415 |
5.46 |
|
|
|
| 26 |
Bu |
1323 |
1274 |
1.70 |
|
|
|
| 27 |
Bu |
1223 |
1177 |
16.89 |
|
|
|
| 28 |
Bu |
1018 |
980 |
1.21 |
|
|
|
| 29 |
Bu |
524 |
505 |
12.04 |
|
|
|
| 30 |
Bu |
109 |
105 |
2.99 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20814.1 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 20033.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.153 |
|
|
|
| 2 |
C |
0.153 |
|
|
|
| 3 |
C |
-0.237 |
|
|
|
| 4 |
C |
-0.237 |
|
|
|
| 5 |
C |
-0.412 |
|
|
|
| 6 |
C |
-0.412 |
|
|
|
| 7 |
H |
0.174 |
|
|
|
| 8 |
H |
0.174 |
|
|
|
| 9 |
H |
0.163 |
|
|
|
| 10 |
H |
0.158 |
|
|
|
| 11 |
H |
0.163 |
|
|
|
| 12 |
H |
0.158 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.348 |
4.863 |
0.000 |
| y |
4.863 |
22.228 |
0.000 |
| z |
0.000 |
0.000 |
6.052 |
<r2> (average value of r
2) Å
2
| <r2> |
250.217 |
| (<r2>)1/2 |
15.818 |