Vibrational Frequencies calculated at TPSSh/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3236 |
3114 |
20.76 |
|
|
|
| 2 |
A' |
3212 |
3091 |
7.01 |
|
|
|
| 3 |
A' |
3205 |
3085 |
28.52 |
|
|
|
| 4 |
A' |
3193 |
3073 |
38.19 |
|
|
|
| 5 |
A' |
3181 |
3061 |
4.68 |
|
|
|
| 6 |
A' |
3174 |
3055 |
5.29 |
|
|
|
| 7 |
A' |
3031 |
2918 |
17.00 |
|
|
|
| 8 |
A' |
1651 |
1589 |
2.11 |
|
|
|
| 9 |
A' |
1638 |
1576 |
0.52 |
|
|
|
| 10 |
A' |
1603 |
1543 |
1.35 |
|
|
|
| 11 |
A' |
1495 |
1439 |
6.73 |
|
|
|
| 12 |
A' |
1492 |
1436 |
4.04 |
|
|
|
| 13 |
A' |
1447 |
1392 |
10.58 |
|
|
|
| 14 |
A' |
1407 |
1354 |
1.22 |
|
|
|
| 15 |
A' |
1360 |
1309 |
2.95 |
|
|
|
| 16 |
A' |
1320 |
1270 |
0.34 |
|
|
|
| 17 |
A' |
1253 |
1206 |
3.16 |
|
|
|
| 18 |
A' |
1233 |
1187 |
2.01 |
|
|
|
| 19 |
A' |
1186 |
1142 |
1.53 |
|
|
|
| 20 |
A' |
1179 |
1135 |
0.20 |
|
|
|
| 21 |
A' |
1137 |
1094 |
0.47 |
|
|
|
| 22 |
A' |
1088 |
1047 |
1.40 |
|
|
|
| 23 |
A' |
1043 |
1004 |
4.69 |
|
|
|
| 24 |
A' |
961 |
925 |
10.86 |
|
|
|
| 25 |
A' |
866 |
833 |
2.83 |
|
|
|
| 26 |
A' |
836 |
805 |
1.54 |
|
|
|
| 27 |
A' |
735 |
707 |
2.30 |
|
|
|
| 28 |
A' |
594 |
571 |
1.79 |
|
|
|
| 29 |
A' |
536 |
516 |
0.15 |
|
|
|
| 30 |
A' |
383 |
369 |
1.47 |
|
|
|
| 31 |
A" |
3061 |
2946 |
8.51 |
|
|
|
| 32 |
A" |
1144 |
1101 |
2.37 |
|
|
|
| 33 |
A" |
987 |
950 |
0.03 |
|
|
|
| 34 |
A" |
960 |
924 |
0.94 |
|
|
|
| 35 |
A" |
953 |
917 |
1.63 |
|
|
|
| 36 |
A" |
933 |
898 |
6.27 |
|
|
|
| 37 |
A" |
871 |
839 |
0.04 |
|
|
|
| 38 |
A" |
781 |
752 |
66.94 |
|
|
|
| 39 |
A" |
729 |
702 |
21.20 |
|
|
|
| 40 |
A" |
705 |
679 |
20.41 |
|
|
|
| 41 |
A" |
557 |
536 |
5.74 |
|
|
|
| 42 |
A" |
421 |
405 |
8.37 |
|
|
|
| 43 |
A" |
389 |
375 |
3.92 |
|
|
|
| 44 |
A" |
209 |
201 |
4.11 |
|
|
|
| 45 |
A" |
192 |
185 |
0.62 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30782.1 cm
-1
Scaled (by 0.9625) Zero Point Vibrational Energy (zpe) 29627.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
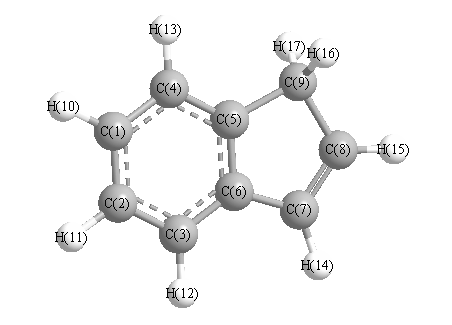
Charges from optimized geometry at TPSSh/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.073 |
|
|
|
| 2 |
C |
-0.133 |
|
|
|
| 3 |
C |
0.095 |
|
|
|
| 4 |
C |
-0.262 |
|
|
|
| 5 |
C |
-0.113 |
|
|
|
| 6 |
C |
0.344 |
|
|
|
| 7 |
C |
-0.658 |
|
|
|
| 8 |
C |
0.173 |
|
|
|
| 9 |
C |
-0.625 |
|
|
|
| 10 |
H |
0.146 |
|
|
|
| 11 |
H |
0.146 |
|
|
|
| 12 |
H |
0.149 |
|
|
|
| 13 |
H |
0.147 |
|
|
|
| 14 |
H |
0.151 |
|
|
|
| 15 |
H |
0.151 |
|
|
|
| 16 |
H |
0.181 |
|
|
|
| 17 |
H |
0.181 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.434 |
0.605 |
0.000 |
0.744 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
19.875 |
1.772 |
0.000 |
| y |
1.772 |
16.738 |
0.000 |
| z |
0.000 |
0.000 |
8.684 |
<r2> (average value of r
2) Å
2
| <r2> |
295.028 |
| (<r2>)1/2 |
17.176 |