Vibrational Frequencies calculated at TPSSh/cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3499 |
3388 |
2.54 |
|
|
|
| 2 |
A |
3362 |
3255 |
330.23 |
|
|
|
| 3 |
A |
3108 |
3009 |
23.51 |
|
|
|
| 4 |
A |
3075 |
2978 |
3.66 |
|
|
|
| 5 |
A |
3069 |
2972 |
31.34 |
|
|
|
| 6 |
A |
3029 |
2933 |
21.22 |
|
|
|
| 7 |
A |
2984 |
2889 |
74.29 |
|
|
|
| 8 |
A |
1839 |
1781 |
365.79 |
|
|
|
| 9 |
A |
1526 |
1478 |
8.60 |
|
|
|
| 10 |
A |
1506 |
1459 |
13.76 |
|
|
|
| 11 |
A |
1489 |
1442 |
14.45 |
|
|
|
| 12 |
A |
1474 |
1427 |
6.36 |
|
|
|
| 13 |
A |
1461 |
1415 |
3.61 |
|
|
|
| 14 |
A |
1418 |
1373 |
361.04 |
|
|
|
| 15 |
A |
1340 |
1297 |
7.67 |
|
|
|
| 16 |
A |
1287 |
1246 |
4.08 |
|
|
|
| 17 |
A |
1212 |
1174 |
20.91 |
|
|
|
| 18 |
A |
1167 |
1130 |
15.29 |
|
|
|
| 19 |
A |
1142 |
1106 |
36.54 |
|
|
|
| 20 |
A |
1123 |
1087 |
25.39 |
|
|
|
| 21 |
A |
1001 |
970 |
21.82 |
|
|
|
| 22 |
A |
957 |
926 |
15.06 |
|
|
|
| 23 |
A |
911 |
882 |
48.07 |
|
|
|
| 24 |
A |
867 |
839 |
19.89 |
|
|
|
| 25 |
A |
778 |
753 |
57.36 |
|
|
|
| 26 |
A |
634 |
614 |
3.01 |
|
|
|
| 27 |
A |
569 |
551 |
6.79 |
|
|
|
| 28 |
A |
471 |
457 |
9.18 |
|
|
|
| 29 |
A |
370 |
358 |
5.41 |
|
|
|
| 30 |
A |
281 |
272 |
5.34 |
|
|
|
| 31 |
A |
202 |
196 |
2.04 |
|
|
|
| 32 |
A |
136 |
132 |
2.52 |
|
|
|
| 33 |
A |
70 |
68 |
5.57 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23678.5 cm
-1
Scaled (by 0.9683) Zero Point Vibrational Energy (zpe) 22927.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
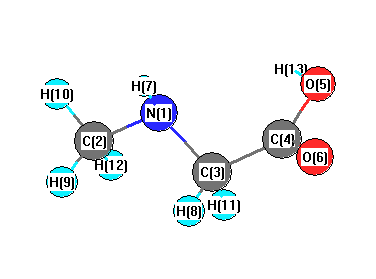
Charges from optimized geometry at TPSSh/cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.246 |
|
|
|
| 2 |
C |
-0.134 |
|
|
|
| 3 |
C |
-0.066 |
|
|
|
| 4 |
C |
0.261 |
|
|
|
| 5 |
O |
-0.238 |
|
|
|
| 6 |
O |
-0.306 |
|
|
|
| 7 |
H |
0.123 |
|
|
|
| 8 |
H |
0.081 |
|
|
|
| 9 |
H |
0.063 |
|
|
|
| 10 |
H |
0.085 |
|
|
|
| 11 |
H |
0.088 |
|
|
|
| 12 |
H |
0.083 |
|
|
|
| 13 |
H |
0.207 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
5.391 |
-0.216 |
-0.650 |
5.434 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.500 |
0.335 |
0.032 |
| y |
0.335 |
7.466 |
0.014 |
| z |
0.032 |
0.014 |
5.903 |
<r2> (average value of r
2) Å
2
| <r2> |
193.201 |
| (<r2>)1/2 |
13.900 |