Jump to
S1C2
S1C3
Energy calculated at TPSSh/3-21G*
| | hartrees |
|---|
| Energy at 0K | -253.625107 |
| Energy at 298.15K | -253.624703 |
| HF Energy | -253.625107 |
| Nuclear repulsion energy | 46.957342 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at TPSSh/3-21G*
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-0.653 |
| N2 |
0.000 |
0.000 |
-1.836 |
| Na3 |
0.000 |
0.000 |
1.525 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.1824 | 2.1781 |
N2 | 1.1824 | | 3.3605 | Na3 | 2.1781 | 3.3605 | |
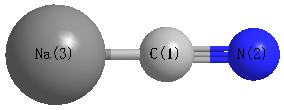 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
0.000 |
|
C1 |
Na3 |
N2 |
0.000 |
| N2 |
C1 |
Na3 |
180.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.147 |
|
|
|
| 2 |
N |
-0.451 |
|
|
|
| 3 |
Na |
0.598 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
9.825 |
9.825 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.805 |
0.000 |
0.000 |
| y |
0.000 |
2.805 |
0.000 |
| z |
0.000 |
0.000 |
5.521 |
<r2> (average value of r
2) Å
2
| <r2> |
61.724 |
| (<r2>)1/2 |
7.856 |
Jump to
S1C1
S1C3
Energy calculated at TPSSh/3-21G*
| | hartrees |
|---|
| Energy at 0K | -253.628684 |
| Energy at 298.15K | -253.628371 |
| HF Energy | -253.628684 |
| Nuclear repulsion energy | 51.650817 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at TPSSh/3-21G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
1.132 |
0.664 |
0.000 |
| N2 |
0.000 |
1.054 |
0.000 |
| Na3 |
-0.618 |
-1.033 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.1975 | 2.4381 |
N2 | 1.1975 | | 2.1765 | Na3 | 2.4381 | 2.1765 | |
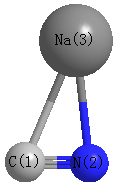 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
87.505 |
|
C1 |
Na3 |
N2 |
29.386 |
| N2 |
C1 |
Na3 |
63.109 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.018 |
|
|
|
| 2 |
N |
-0.501 |
|
|
|
| 3 |
Na |
0.484 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-4.642 |
-7.121 |
0.000 |
8.500 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.069 |
0.118 |
0.000 |
| y |
0.118 |
3.337 |
0.000 |
| z |
0.000 |
0.000 |
2.852 |
<r2> (average value of r
2) Å
2
| <r2> |
45.288 |
| (<r2>)1/2 |
6.730 |
Jump to
S1C1
S1C2
Energy calculated at TPSSh/3-21G*
| | hartrees |
|---|
| Energy at 0K | -253.627787 |
| Energy at 298.15K | -253.626999 |
| HF Energy | -253.627787 |
| Nuclear repulsion energy | 49.352844 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Geometric Data calculated at TPSSh/3-21G*
Point Group is C∞v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.832 |
| N2 |
0.000 |
0.000 |
-0.638 |
| Na3 |
0.000 |
0.000 |
1.405 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
Na3 |
| C1 | | 1.1933 | 3.2370 |
N2 | 1.1933 | | 2.0437 | Na3 | 3.2370 | 2.0437 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
Na3 |
180.000 |
|
C1 |
Na3 |
N2 |
0.000 |
| N2 |
C1 |
Na3 |
0.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at TPSSh/3-21G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.040 |
|
|
|
| 2 |
N |
-0.599 |
|
|
|
| 3 |
Na |
0.559 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
9.916 |
9.916 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.667 |
0.000 |
0.000 |
| y |
0.000 |
2.667 |
0.000 |
| z |
0.000 |
0.000 |
5.685 |
<r2> (average value of r
2) Å
2
| <r2> |
55.175 |
| (<r2>)1/2 |
7.428 |