Jump to
S1C2
Energy calculated at B3LYP/6-31G*
| | hartrees |
|---|
| Energy at 0K | -2610.944769 |
| Energy at 298.15K | -2610.948566 |
| HF Energy | -2610.944769 |
| Nuclear repulsion energy | 80.179235 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3196 |
3070 |
5.96 |
|
|
|
| 2 |
A' |
1406 |
1350 |
23.92 |
|
|
|
| 3 |
A' |
691 |
663 |
20.80 |
|
|
|
| 4 |
A' |
274 |
263 |
71.15 |
|
|
|
| 5 |
A" |
3347 |
3214 |
0.09 |
|
|
|
| 6 |
A" |
934 |
897 |
1.34 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 4923.9 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 4728.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G*
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-0.006 |
1.497 |
0.000 |
| Br2 |
-0.006 |
-0.370 |
0.000 |
| H3 |
0.126 |
1.991 |
0.952 |
| H4 |
0.126 |
1.991 |
-0.952 |
Atom - Atom Distances (Å)
| |
C1 |
Br2 |
H3 |
H4 |
| C1 | | 1.8678 | 1.0807 | 1.0807 |
Br2 | 1.8678 | | 2.5497 | 2.5497 | H3 | 1.0807 | 2.5497 | | 1.9044 | H4 | 1.0807 | 2.5497 | 1.9044 | |
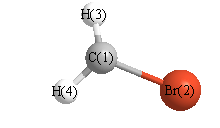 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Br2 |
C1 |
H3 |
117.184 |
|
Br2 |
C1 |
H4 |
117.184 |
| H3 |
C1 |
H4 |
123.546 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.327 |
|
|
|
| 2 |
Br |
-0.055 |
|
|
|
| 3 |
H |
0.191 |
|
|
|
| 4 |
H |
0.191 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.239 |
1.008 |
0.000 |
1.036 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.888 |
0.508 |
0.000 |
| y |
0.508 |
-21.412 |
0.000 |
| z |
0.000 |
0.000 |
-24.073 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.145 |
0.508 |
0.000 |
| y |
0.508 |
3.568 |
0.000 |
| z |
0.000 |
0.000 |
-0.424 |
|
| Polar |
|---|
| 3z2-r2 | -0.847 |
|---|
| x2-y2 | -4.475 |
|---|
| xy | 0.508 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.377 |
0.062 |
0.000 |
| y |
0.062 |
5.470 |
0.000 |
| z |
0.000 |
0.000 |
3.156 |
<r2> (average value of r
2) Å
2
| <r2> |
42.890 |
| (<r2>)1/2 |
6.549 |
Jump to
S1C1
Energy calculated at B3LYP/6-31G*
| | hartrees |
|---|
| Energy at 0K | -2610.944731 |
| Energy at 298.15K | |
| HF Energy | -2610.944731 |
| Nuclear repulsion energy | 80.220607 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3203 |
3076 |
4.70 |
107.24 |
0.12 |
0.22 |
| 2 |
A1 |
1403 |
1348 |
24.77 |
2.50 |
0.66 |
0.80 |
| 3 |
A1 |
688 |
661 |
19.55 |
9.24 |
0.23 |
0.37 |
| 4 |
B1 |
197i |
190i |
80.75 |
0.46 |
0.75 |
0.86 |
| 5 |
B2 |
3357 |
3223 |
0.00 |
59.10 |
0.75 |
0.86 |
| 6 |
B2 |
928 |
891 |
1.46 |
4.38 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 4690.4 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 4504.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G*
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
-1.495 |
| Br2 |
0.000 |
0.000 |
0.371 |
| H3 |
0.000 |
0.956 |
-1.998 |
| H4 |
0.000 |
-0.956 |
-1.998 |
Atom - Atom Distances (Å)
| |
C1 |
Br2 |
H3 |
H4 |
| C1 | | 1.8658 | 1.0800 | 1.0800 |
Br2 | 1.8658 | | 2.5543 | 2.5543 | H3 | 1.0800 | 2.5543 | | 1.9113 | H4 | 1.0800 | 2.5543 | 1.9113 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Br2 |
C1 |
H3 |
117.759 |
|
Br2 |
C1 |
H4 |
117.759 |
| H3 |
C1 |
H4 |
124.481 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.331 |
|
|
|
| 2 |
Br |
-0.053 |
|
|
|
| 3 |
H |
0.192 |
|
|
|
| 4 |
H |
0.192 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-1.016 |
1.016 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-25.931 |
0.000 |
0.000 |
| y |
0.000 |
-24.037 |
0.000 |
| z |
0.000 |
0.000 |
-21.342 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.241 |
0.000 |
0.000 |
| y |
0.000 |
-0.400 |
0.000 |
| z |
0.000 |
0.000 |
3.641 |
|
| Polar |
|---|
| 3z2-r2 | 7.282 |
|---|
| x2-y2 | -1.894 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
2.359 |
0.000 |
0.000 |
| y |
0.000 |
3.147 |
0.000 |
| z |
0.000 |
0.000 |
5.453 |
<r2> (average value of r
2) Å
2
| <r2> |
42.878 |
| (<r2>)1/2 |
6.548 |