Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3213 |
3085 |
0.00 |
|
|
|
| 2 |
Ag |
3197 |
3070 |
0.00 |
|
|
|
| 3 |
Ag |
1722 |
1653 |
0.00 |
|
|
|
| 4 |
Ag |
1521 |
1460 |
0.00 |
|
|
|
| 5 |
Ag |
1450 |
1393 |
0.00 |
|
|
|
| 6 |
Ag |
1201 |
1153 |
0.00 |
|
|
|
| 7 |
Ag |
1143 |
1097 |
0.00 |
|
|
|
| 8 |
Ag |
1023 |
982 |
0.00 |
|
|
|
| 9 |
Ag |
784 |
752 |
0.00 |
|
|
|
| 10 |
Ag |
402 |
386 |
0.00 |
|
|
|
| 11 |
Au |
971 |
932 |
0.00 |
|
|
|
| 12 |
Au |
903 |
867 |
0.00 |
|
|
|
| 13 |
Au |
825 |
793 |
0.00 |
|
|
|
| 14 |
Au |
586 |
563 |
0.00 |
|
|
|
| 15 |
Au |
150 |
144 |
0.00 |
|
|
|
| 16 |
B1g |
971 |
933 |
0.00 |
|
|
|
| 17 |
B1g |
885 |
850 |
0.00 |
|
|
|
| 18 |
B1g |
719 |
691 |
0.00 |
|
|
|
| 19 |
B1g |
467 |
448 |
0.00 |
|
|
|
| 20 |
B1u |
3211 |
3084 |
52.58 |
|
|
|
| 21 |
B1u |
3196 |
3069 |
21.22 |
|
|
|
| 22 |
B1u |
1649 |
1584 |
0.49 |
|
|
|
| 23 |
B1u |
1478 |
1420 |
32.25 |
|
|
|
| 24 |
B1u |
1318 |
1265 |
31.11 |
|
|
|
| 25 |
B1u |
1192 |
1144 |
10.01 |
|
|
|
| 26 |
B1u |
1057 |
1015 |
0.00 |
|
|
|
| 27 |
B1u |
999 |
960 |
17.79 |
|
|
|
| 28 |
B1u |
628 |
603 |
4.26 |
|
|
|
| 29 |
B2g |
924 |
887 |
0.00 |
|
|
|
| 30 |
B2g |
763 |
733 |
0.00 |
|
|
|
| 31 |
B2g |
451 |
433 |
0.00 |
|
|
|
| 32 |
B2g |
325 |
312 |
0.00 |
|
|
|
| 33 |
B2u |
3206 |
3079 |
60.71 |
|
|
|
| 34 |
B2u |
3184 |
3057 |
0.60 |
|
|
|
| 35 |
B2u |
1643 |
1577 |
1.69 |
|
|
|
| 36 |
B2u |
1493 |
1434 |
5.18 |
|
|
|
| 37 |
B2u |
1305 |
1254 |
2.01 |
|
|
|
| 38 |
B2u |
1156 |
1110 |
10.50 |
|
|
|
| 39 |
B2u |
1076 |
1033 |
1.21 |
|
|
|
| 40 |
B2u |
737 |
708 |
0.34 |
|
|
|
| 41 |
B2u |
215 |
206 |
0.67 |
|
|
|
| 42 |
B3g |
3206 |
3078 |
0.00 |
|
|
|
| 43 |
B3g |
3183 |
3057 |
0.00 |
|
|
|
| 44 |
B3g |
1657 |
1592 |
0.00 |
|
|
|
| 45 |
B3g |
1496 |
1436 |
0.00 |
|
|
|
| 46 |
B3g |
1322 |
1269 |
0.00 |
|
|
|
| 47 |
B3g |
1120 |
1075 |
0.00 |
|
|
|
| 48 |
B3g |
1001 |
962 |
0.00 |
|
|
|
| 49 |
B3g |
615 |
591 |
0.00 |
|
|
|
| 50 |
B3g |
568 |
545 |
0.00 |
|
|
|
| 51 |
B3u |
919 |
883 |
6.17 |
|
|
|
| 52 |
B3u |
751 |
721 |
103.07 |
|
|
|
| 53 |
B3u |
382 |
367 |
1.42 |
|
|
|
| 54 |
B3u |
108 |
104 |
1.87 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34831.8 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 33449.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
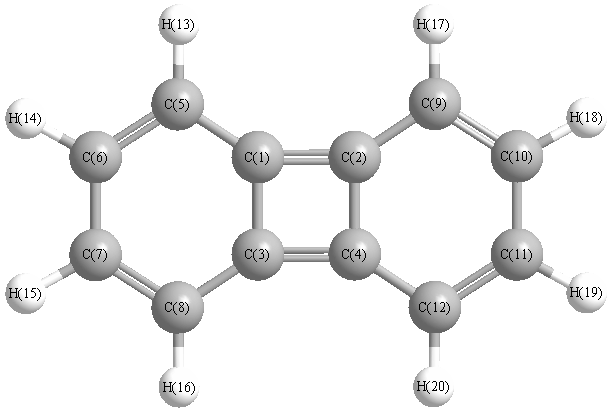
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.113 |
|
|
|
| 2 |
C |
0.113 |
|
|
|
| 3 |
C |
0.113 |
|
|
|
| 4 |
C |
0.113 |
|
|
|
| 5 |
C |
-0.245 |
|
|
|
| 6 |
C |
-0.136 |
|
|
|
| 7 |
C |
-0.136 |
|
|
|
| 8 |
C |
-0.245 |
|
|
|
| 9 |
C |
-0.245 |
|
|
|
| 10 |
C |
-0.136 |
|
|
|
| 11 |
C |
-0.136 |
|
|
|
| 12 |
C |
-0.245 |
|
|
|
| 13 |
H |
0.137 |
|
|
|
| 14 |
H |
0.130 |
|
|
|
| 15 |
H |
0.130 |
|
|
|
| 16 |
H |
0.137 |
|
|
|
| 17 |
H |
0.137 |
|
|
|
| 18 |
H |
0.130 |
|
|
|
| 19 |
H |
0.130 |
|
|
|
| 20 |
H |
0.137 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-73.003 |
0.000 |
0.000 |
| y |
0.000 |
-60.013 |
0.000 |
| z |
0.000 |
0.000 |
-59.420 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-13.286 |
0.000 |
0.000 |
| y |
0.000 |
6.199 |
0.000 |
| z |
0.000 |
0.000 |
7.088 |
|
| Polar |
|---|
| 3z2-r2 | 14.175 |
|---|
| x2-y2 | -12.990 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.715 |
0.000 |
0.000 |
| y |
0.000 |
18.621 |
0.000 |
| z |
0.000 |
0.000 |
30.784 |
<r2> (average value of r
2) Å
2
| <r2> |
562.473 |
| (<r2>)1/2 |
23.717 |