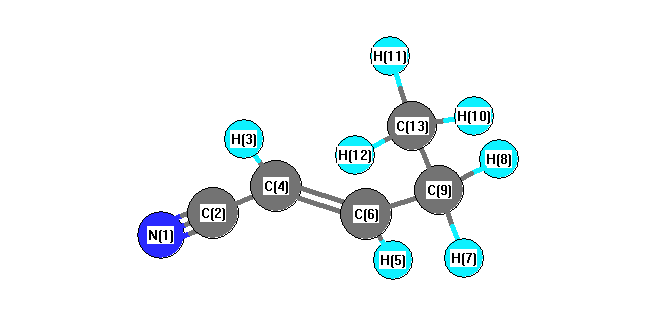
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3202 |
3075 |
2.90 |
|
|
|
| 2 |
A' |
3174 |
3048 |
8.21 |
|
|
|
| 3 |
A' |
3129 |
3005 |
25.95 |
|
|
|
| 4 |
A' |
3056 |
2934 |
17.46 |
|
|
|
| 5 |
A' |
3019 |
2899 |
18.06 |
|
|
|
| 6 |
A' |
2344 |
2251 |
25.75 |
|
|
|
| 7 |
A' |
1711 |
1643 |
26.82 |
|
|
|
| 8 |
A' |
1537 |
1476 |
4.96 |
|
|
|
| 9 |
A' |
1496 |
1437 |
9.20 |
|
|
|
| 10 |
A' |
1447 |
1389 |
4.71 |
|
|
|
| 11 |
A' |
1401 |
1345 |
10.65 |
|
|
|
| 12 |
A' |
1349 |
1295 |
1.12 |
|
|
|
| 13 |
A' |
1314 |
1261 |
2.28 |
|
|
|
| 14 |
A' |
1119 |
1075 |
1.94 |
|
|
|
| 15 |
A' |
1075 |
1032 |
4.81 |
|
|
|
| 16 |
A' |
998 |
958 |
1.71 |
|
|
|
| 17 |
A' |
900 |
864 |
3.45 |
|
|
|
| 18 |
A' |
578 |
555 |
0.49 |
|
|
|
| 19 |
A' |
510 |
489 |
0.04 |
|
|
|
| 20 |
A' |
260 |
250 |
1.39 |
|
|
|
| 21 |
A' |
148 |
142 |
4.18 |
|
|
|
| 22 |
A" |
3122 |
2998 |
28.81 |
|
|
|
| 23 |
A" |
3038 |
2918 |
12.76 |
|
|
|
| 24 |
A" |
1531 |
1471 |
7.13 |
|
|
|
| 25 |
A" |
1303 |
1252 |
0.15 |
|
|
|
| 26 |
A" |
1116 |
1072 |
1.11 |
|
|
|
| 27 |
A" |
1006 |
966 |
26.05 |
|
|
|
| 28 |
A" |
856 |
822 |
8.28 |
|
|
|
| 29 |
A" |
744 |
715 |
0.30 |
|
|
|
| 30 |
A" |
494 |
475 |
6.29 |
|
|
|
| 31 |
A" |
280 |
269 |
0.00 |
|
|
|
| 32 |
A" |
178 |
171 |
0.00 |
|
|
|
| 33 |
A" |
123 |
119 |
3.93 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23778.5 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 22834.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.477 |
-0.470 |
|
-0.459 |
| 2 |
C |
0.308 |
0.420 |
|
0.417 |
| 3 |
H |
0.178 |
0.162 |
|
0.185 |
| 4 |
C |
-0.149 |
-0.283 |
|
-0.327 |
| 5 |
H |
0.161 |
0.101 |
|
0.112 |
| 6 |
C |
-0.067 |
-0.068 |
|
-0.068 |
| 7 |
H |
0.166 |
-0.000 |
|
0.015 |
| 8 |
H |
0.166 |
-0.000 |
|
0.015 |
| 9 |
C |
-0.303 |
0.170 |
|
0.144 |
| 10 |
H |
0.157 |
0.048 |
|
0.065 |
| 11 |
H |
0.158 |
0.046 |
|
0.068 |
| 12 |
H |
0.158 |
0.046 |
|
0.068 |
| 13 |
C |
-0.456 |
-0.172 |
|
-0.234 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.116 |
-4.320 |
0.000 |
4.810 |
| CHELPG |
2.109 |
-4.262 |
0.000 |
4.755 |
| AIM |
|
|
|
|
| ESP |
2.129 |
-4.298 |
0.000 |
4.797 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.696 |
6.950 |
0.000 |
| y |
6.950 |
-44.942 |
0.000 |
| z |
0.000 |
0.000 |
-36.736 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.143 |
6.950 |
0.000 |
| y |
6.950 |
-7.726 |
0.000 |
| z |
0.000 |
0.000 |
4.583 |
|
| Polar |
|---|
| 3z2-r2 | 9.166 |
|---|
| x2-y2 | 7.246 |
|---|
| xy | 6.950 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.798 |
-1.853 |
0.000 |
| y |
-1.853 |
12.470 |
0.000 |
| z |
0.000 |
0.000 |
5.042 |
<r2> (average value of r
2) Å
2
| <r2> |
227.795 |
| (<r2>)1/2 |
15.093 |