Jump to
S1C2
Energy calculated at B3LYP/6-31G*
| | hartrees |
|---|
| Energy at 0K | -345.329814 |
| Energy at 298.15K | -345.346981 |
| Nuclear repulsion energy | 423.615511 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1' |
3056 |
2935 |
0.00 |
|
|
|
| 2 |
A1' |
1527 |
1466 |
0.00 |
|
|
|
| 3 |
A1' |
1378 |
1323 |
0.00 |
|
|
|
| 4 |
A1' |
967 |
928 |
0.00 |
|
|
|
| 5 |
A1' |
813 |
781 |
0.00 |
|
|
|
| 6 |
A1' |
608 |
584 |
0.00 |
|
|
|
| 7 |
A1" |
3075 |
2952 |
0.00 |
|
|
|
| 8 |
A1" |
1277 |
1227 |
0.00 |
|
|
|
| 9 |
A1" |
1038 |
996 |
0.00 |
|
|
|
| 10 |
A1" |
107 |
103 |
0.00 |
|
|
|
| 11 |
A2' |
3095 |
2973 |
0.00 |
|
|
|
| 12 |
A2' |
1208 |
1160 |
0.00 |
|
|
|
| 13 |
A2' |
819 |
786 |
0.00 |
|
|
|
| 14 |
A2" |
3042 |
2921 |
122.83 |
|
|
|
| 15 |
A2" |
1520 |
1460 |
2.40 |
|
|
|
| 16 |
A2" |
1403 |
1347 |
10.07 |
|
|
|
| 17 |
A2" |
999 |
959 |
20.43 |
|
|
|
| 18 |
A2" |
767 |
737 |
61.37 |
|
|
|
| 19 |
E' |
3103 |
2980 |
101.21 |
|
|
|
| 19 |
E' |
3103 |
2980 |
101.22 |
|
|
|
| 20 |
E' |
3047 |
2926 |
109.61 |
|
|
|
| 20 |
E' |
3047 |
2926 |
109.64 |
|
|
|
| 21 |
E' |
1519 |
1458 |
6.86 |
|
|
|
| 21 |
E' |
1519 |
1458 |
6.85 |
|
|
|
| 22 |
E' |
1365 |
1311 |
7.65 |
|
|
|
| 22 |
E' |
1365 |
1311 |
7.65 |
|
|
|
| 23 |
E' |
1347 |
1293 |
0.00 |
|
|
|
| 23 |
E' |
1347 |
1293 |
0.00 |
|
|
|
| 24 |
E' |
1083 |
1040 |
41.36 |
|
|
|
| 24 |
E' |
1083 |
1040 |
41.36 |
|
|
|
| 25 |
E' |
899 |
863 |
3.61 |
|
|
|
| 25 |
E' |
899 |
863 |
3.61 |
|
|
|
| 26 |
E' |
836 |
803 |
4.63 |
|
|
|
| 26 |
E' |
836 |
803 |
4.63 |
|
|
|
| 27 |
E' |
428 |
411 |
0.00 |
|
|
|
| 27 |
E' |
428 |
411 |
0.00 |
|
|
|
| 28 |
E" |
3078 |
2956 |
0.00 |
|
|
|
| 28 |
E" |
3078 |
2956 |
0.00 |
|
|
|
| 29 |
E" |
3039 |
2918 |
0.00 |
|
|
|
| 29 |
E" |
3039 |
2918 |
0.00 |
|
|
|
| 30 |
E" |
1508 |
1448 |
0.00 |
|
|
|
| 30 |
E" |
1508 |
1448 |
0.00 |
|
|
|
| 31 |
E" |
1360 |
1306 |
0.00 |
|
|
|
| 31 |
E" |
1360 |
1306 |
0.00 |
|
|
|
| 32 |
E" |
1342 |
1289 |
0.00 |
|
|
|
| 32 |
E" |
1342 |
1289 |
0.00 |
|
|
|
| 33 |
E" |
1216 |
1168 |
0.00 |
|
|
|
| 33 |
E" |
1216 |
1168 |
0.00 |
|
|
|
| 34 |
E" |
1045 |
1003 |
0.00 |
|
|
|
| 34 |
E" |
1045 |
1003 |
0.00 |
|
|
|
| 35 |
E" |
591 |
567 |
0.00 |
|
|
|
| 35 |
E" |
591 |
567 |
0.00 |
|
|
|
| 36 |
E" |
342 |
328 |
0.00 |
|
|
|
| 36 |
E" |
342 |
328 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40495.6 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 38887.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G*
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.290 |
| N2 |
0.000 |
0.000 |
-1.290 |
| C3 |
0.000 |
1.384 |
0.783 |
| C4 |
1.198 |
-0.692 |
0.783 |
| C5 |
-1.198 |
-0.692 |
0.783 |
| C6 |
0.000 |
1.384 |
-0.783 |
| C7 |
-1.198 |
-0.692 |
-0.783 |
| C8 |
1.198 |
-0.692 |
-0.783 |
| H9 |
0.884 |
1.893 |
1.185 |
| H10 |
-0.884 |
1.893 |
1.185 |
| H11 |
1.197 |
-1.712 |
1.185 |
| H12 |
2.081 |
-0.181 |
1.185 |
| H13 |
-2.081 |
-0.181 |
1.185 |
| H14 |
-1.197 |
-1.712 |
1.185 |
| H15 |
-0.884 |
1.893 |
-1.185 |
| H16 |
0.884 |
1.893 |
-1.185 |
| H17 |
-1.197 |
-1.712 |
-1.185 |
| H18 |
-2.081 |
-0.181 |
-1.185 |
| H19 |
2.081 |
-0.181 |
-1.185 |
| H20 |
1.197 |
-1.712 |
-1.185 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5806 | 1.4737 | 1.4737 | 1.4737 | 2.4923 | 2.4923 | 2.4923 | 2.0916 | 2.0916 | 2.0916 | 2.0916 | 2.0916 | 2.0916 | 3.2393 | 3.2393 | 3.2393 | 3.2393 | 3.2393 | 3.2393 |
N2 | 2.5806 | | 2.4923 | 2.4923 | 2.4923 | 1.4737 | 1.4737 | 1.4737 | 3.2393 | 3.2393 | 3.2393 | 3.2393 | 3.2393 | 3.2393 | 2.0916 | 2.0916 | 2.0916 | 2.0916 | 2.0916 | 2.0916 | C3 | 1.4737 | 2.4923 | | 2.3964 | 2.3964 | 1.5654 | 2.8624 | 2.8624 | 1.0967 | 1.0967 | 3.3433 | 2.6346 | 2.6346 | 3.3433 | 2.2167 | 2.2167 | 3.8586 | 3.2637 | 3.2637 | 3.8586 | C4 | 1.4737 | 2.4923 | 2.3964 | | 2.3964 | 2.8624 | 2.8624 | 1.5654 | 2.6346 | 3.3433 | 1.0967 | 1.0967 | 3.3433 | 2.6346 | 3.8586 | 3.2638 | 3.2637 | 3.8586 | 2.2167 | 2.2167 | C5 | 1.4737 | 2.4923 | 2.3964 | 2.3964 | | 2.8624 | 1.5654 | 2.8624 | 3.3433 | 2.6346 | 2.6346 | 3.3433 | 1.0967 | 1.0967 | 3.2638 | 3.8586 | 2.2167 | 2.2167 | 3.8586 | 3.2637 | C6 | 2.4923 | 1.4737 | 1.5654 | 2.8624 | 2.8624 | | 2.3964 | 2.3964 | 2.2167 | 2.2167 | 3.8586 | 3.2637 | 3.2637 | 3.8586 | 1.0967 | 1.0967 | 3.3433 | 2.6346 | 2.6346 | 3.3433 | C7 | 2.4923 | 1.4737 | 2.8624 | 2.8624 | 1.5654 | 2.3964 | | 2.3964 | 3.8586 | 3.2638 | 3.2637 | 3.8586 | 2.2167 | 2.2167 | 2.6346 | 3.3433 | 1.0967 | 1.0967 | 3.3433 | 2.6346 | C8 | 2.4923 | 1.4737 | 2.8624 | 1.5654 | 2.8624 | 2.3964 | 2.3964 | | 3.2638 | 3.8586 | 2.2167 | 2.2167 | 3.8586 | 3.2637 | 3.3433 | 2.6346 | 2.6346 | 3.3433 | 1.0967 | 1.0967 | H9 | 2.0916 | 3.2393 | 1.0967 | 2.6346 | 3.3433 | 2.2167 | 3.8586 | 3.2638 | | 1.7678 | 3.6183 | 2.3945 | 3.6183 | 4.1623 | 2.9573 | 2.3707 | 4.7901 | 4.3258 | 3.3696 | 4.3258 | H10 | 2.0916 | 3.2393 | 1.0967 | 3.3433 | 2.6346 | 2.2167 | 3.2638 | 3.8586 | 1.7678 | | 4.1623 | 3.6183 | 2.3945 | 3.6183 | 2.3707 | 2.9573 | 4.3258 | 3.3696 | 4.3258 | 4.7901 | H11 | 2.0916 | 3.2393 | 3.3433 | 1.0967 | 2.6346 | 3.8586 | 3.2637 | 2.2167 | 3.6183 | 4.1623 | | 1.7678 | 3.6183 | 2.3945 | 4.7901 | 4.3258 | 3.3696 | 4.3258 | 2.9573 | 2.3707 | H12 | 2.0916 | 3.2393 | 2.6346 | 1.0967 | 3.3433 | 3.2637 | 3.8586 | 2.2167 | 2.3945 | 3.6183 | 1.7678 | | 4.1623 | 3.6183 | 4.3258 | 3.3696 | 4.3258 | 4.7901 | 2.3707 | 2.9573 | H13 | 2.0916 | 3.2393 | 2.6346 | 3.3433 | 1.0967 | 3.2637 | 2.2167 | 3.8586 | 3.6183 | 2.3945 | 3.6183 | 4.1623 | | 1.7678 | 3.3696 | 4.3258 | 2.9573 | 2.3707 | 4.7901 | 4.3258 | H14 | 2.0916 | 3.2393 | 3.3433 | 2.6346 | 1.0967 | 3.8586 | 2.2167 | 3.2637 | 4.1623 | 3.6183 | 2.3945 | 3.6183 | 1.7678 | | 4.3258 | 4.7901 | 2.3707 | 2.9573 | 4.3258 | 3.3696 | H15 | 3.2393 | 2.0916 | 2.2167 | 3.8586 | 3.2638 | 1.0967 | 2.6346 | 3.3433 | 2.9573 | 2.3707 | 4.7901 | 4.3258 | 3.3696 | 4.3258 | | 1.7678 | 3.6183 | 2.3945 | 3.6183 | 4.1623 | H16 | 3.2393 | 2.0916 | 2.2167 | 3.2638 | 3.8586 | 1.0967 | 3.3433 | 2.6346 | 2.3707 | 2.9573 | 4.3258 | 3.3696 | 4.3258 | 4.7901 | 1.7678 | | 4.1623 | 3.6183 | 2.3945 | 3.6183 | H17 | 3.2393 | 2.0916 | 3.8586 | 3.2637 | 2.2167 | 3.3433 | 1.0967 | 2.6346 | 4.7901 | 4.3258 | 3.3696 | 4.3258 | 2.9573 | 2.3707 | 3.6183 | 4.1623 | | 1.7678 | 3.6183 | 2.3945 | H18 | 3.2393 | 2.0916 | 3.2637 | 3.8586 | 2.2167 | 2.6346 | 1.0967 | 3.3433 | 4.3258 | 3.3696 | 4.3258 | 4.7901 | 2.3707 | 2.9573 | 2.3945 | 3.6183 | 1.7678 | | 4.1623 | 3.6183 | H19 | 3.2393 | 2.0916 | 3.2637 | 2.2167 | 3.8586 | 2.6346 | 3.3433 | 1.0967 | 3.3696 | 4.3258 | 2.9573 | 2.3707 | 4.7901 | 4.3258 | 3.6183 | 2.3945 | 3.6183 | 4.1623 | | 1.7678 | H20 | 3.2393 | 2.0916 | 3.8586 | 2.2167 | 3.2637 | 3.3433 | 2.6346 | 1.0967 | 4.3258 | 4.7901 | 2.3707 | 2.9573 | 4.3258 | 3.3696 | 4.1623 | 3.6183 | 2.3945 | 3.6183 | 1.7678 | |
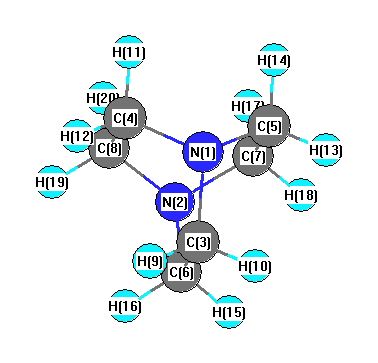 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
110.147 |
|
N1 |
C3 |
H9 |
108.027 |
| N1 |
C3 |
H10 |
108.027 |
|
N1 |
C4 |
C8 |
110.147 |
| N1 |
C4 |
H11 |
108.027 |
|
N1 |
C4 |
H12 |
108.027 |
| N1 |
C5 |
C7 |
110.147 |
|
N1 |
C5 |
H13 |
108.027 |
| N1 |
C5 |
H14 |
108.027 |
|
N2 |
C6 |
C3 |
110.147 |
| N2 |
C6 |
H15 |
108.027 |
|
N2 |
C6 |
H16 |
108.027 |
| N2 |
C7 |
C5 |
110.147 |
|
N2 |
C7 |
H17 |
108.027 |
| N2 |
C7 |
H18 |
108.027 |
|
N2 |
C8 |
C4 |
110.147 |
| N2 |
C8 |
H19 |
108.027 |
|
N2 |
C8 |
H20 |
108.027 |
| C3 |
N1 |
C4 |
108.787 |
|
C3 |
N1 |
C5 |
108.787 |
| C3 |
C6 |
H15 |
111.541 |
|
C3 |
C6 |
H16 |
111.541 |
| C4 |
N1 |
C5 |
108.787 |
|
C4 |
C8 |
H19 |
111.541 |
| C4 |
C8 |
H20 |
111.541 |
|
C5 |
C6 |
H15 |
101.542 |
| C5 |
C6 |
H16 |
151.026 |
|
C6 |
N2 |
C7 |
108.787 |
| C6 |
N2 |
C8 |
108.787 |
|
C6 |
C3 |
H9 |
111.541 |
| C6 |
C3 |
H10 |
111.541 |
|
C7 |
N2 |
C8 |
108.787 |
| C7 |
C5 |
H13 |
111.541 |
|
C7 |
C5 |
H14 |
111.541 |
| C8 |
C4 |
H11 |
111.541 |
|
C8 |
C4 |
H12 |
111.541 |
| H9 |
C3 |
H10 |
107.408 |
|
H11 |
C4 |
H12 |
107.408 |
| H13 |
C5 |
H14 |
107.408 |
|
H15 |
C6 |
H16 |
107.408 |
| H17 |
C7 |
H18 |
107.408 |
|
H19 |
C8 |
H20 |
107.408 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.367 |
|
|
|
| 2 |
N |
-0.367 |
|
|
|
| 3 |
C |
-0.157 |
|
|
|
| 4 |
C |
-0.157 |
|
|
|
| 5 |
C |
-0.157 |
|
|
|
| 6 |
C |
-0.157 |
|
|
|
| 7 |
C |
-0.157 |
|
|
|
| 8 |
C |
-0.157 |
|
|
|
| 9 |
H |
0.140 |
|
|
|
| 10 |
H |
0.140 |
|
|
|
| 11 |
H |
0.140 |
|
|
|
| 12 |
H |
0.140 |
|
|
|
| 13 |
H |
0.140 |
|
|
|
| 14 |
H |
0.140 |
|
|
|
| 15 |
H |
0.140 |
|
|
|
| 16 |
H |
0.140 |
|
|
|
| 17 |
H |
0.140 |
|
|
|
| 18 |
H |
0.140 |
|
|
|
| 19 |
H |
0.140 |
|
|
|
| 20 |
H |
0.140 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.726 |
0.000 |
0.000 |
| y |
0.000 |
-46.726 |
0.000 |
| z |
0.000 |
0.000 |
-57.374 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.324 |
0.000 |
0.000 |
| y |
0.000 |
5.324 |
0.000 |
| z |
0.000 |
0.000 |
-10.648 |
|
| Polar |
|---|
| 3z2-r2 | -21.297 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.945 |
0.000 |
0.000 |
| y |
0.000 |
10.945 |
0.000 |
| z |
0.000 |
0.000 |
9.352 |
<r2> (average value of r
2) Å
2
| <r2> |
214.904 |
| (<r2>)1/2 |
14.660 |
Jump to
S1C1
Energy calculated at B3LYP/6-31G*
| | hartrees |
|---|
| Energy at 0K | -345.329805 |
| Energy at 298.15K | -345.346955 |
| HF Energy | -345.329805 |
| Nuclear repulsion energy | 423.621530 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3075 |
2953 |
0.00 |
|
|
|
| 2 |
A1 |
3056 |
2935 |
0.00 |
|
|
|
| 3 |
A1 |
1527 |
1467 |
0.00 |
|
|
|
| 4 |
A1 |
1378 |
1323 |
0.00 |
|
|
|
| 5 |
A1 |
1277 |
1227 |
0.00 |
|
|
|
| 6 |
A1 |
1037 |
996 |
0.00 |
|
|
|
| 7 |
A1 |
967 |
928 |
0.00 |
|
|
|
| 8 |
A1 |
813 |
781 |
0.00 |
|
|
|
| 9 |
A1 |
608 |
584 |
0.00 |
|
|
|
| 10 |
A1 |
100 |
96 |
0.00 |
|
|
|
| 11 |
A2 |
3095 |
2973 |
0.00 |
|
|
|
| 12 |
A2 |
3042 |
2921 |
122.90 |
|
|
|
| 13 |
A2 |
1520 |
1460 |
2.39 |
|
|
|
| 14 |
A2 |
1403 |
1347 |
10.11 |
|
|
|
| 15 |
A2 |
1207 |
1159 |
0.00 |
|
|
|
| 16 |
A2 |
999 |
959 |
20.52 |
|
|
|
| 17 |
A2 |
817 |
785 |
0.00 |
|
|
|
| 18 |
A2 |
767 |
736 |
61.28 |
|
|
|
| 19 |
E |
3103 |
2980 |
101.06 |
|
|
|
| 19 |
E |
3103 |
2980 |
101.07 |
|
|
|
| 20 |
E |
3078 |
2956 |
0.01 |
|
|
|
| 20 |
E |
3078 |
2956 |
0.01 |
|
|
|
| 21 |
E |
3047 |
2926 |
109.76 |
|
|
|
| 21 |
E |
3047 |
2926 |
109.79 |
|
|
|
| 22 |
E |
3039 |
2918 |
0.00 |
|
|
|
| 22 |
E |
3039 |
2918 |
0.00 |
|
|
|
| 23 |
E |
1518 |
1458 |
6.86 |
|
|
|
| 23 |
E |
1518 |
1458 |
6.87 |
|
|
|
| 24 |
E |
1508 |
1448 |
0.00 |
|
|
|
| 24 |
E |
1508 |
1448 |
0.00 |
|
|
|
| 25 |
E |
1365 |
1311 |
7.66 |
|
|
|
| 25 |
E |
1365 |
1311 |
7.65 |
|
|
|
| 26 |
E |
1359 |
1306 |
0.00 |
|
|
|
| 26 |
E |
1359 |
1306 |
0.00 |
|
|
|
| 27 |
E |
1346 |
1293 |
0.00 |
|
|
|
| 27 |
E |
1346 |
1293 |
0.00 |
|
|
|
| 28 |
E |
1342 |
1289 |
0.00 |
|
|
|
| 28 |
E |
1342 |
1289 |
0.00 |
|
|
|
| 29 |
E |
1217 |
1168 |
0.00 |
|
|
|
| 29 |
E |
1217 |
1168 |
0.00 |
|
|
|
| 30 |
E |
1083 |
1040 |
41.34 |
|
|
|
| 30 |
E |
1083 |
1040 |
41.34 |
|
|
|
| 31 |
E |
1044 |
1003 |
0.00 |
|
|
|
| 31 |
E |
1044 |
1003 |
0.00 |
|
|
|
| 32 |
E |
899 |
863 |
3.58 |
|
|
|
| 32 |
E |
899 |
863 |
3.58 |
|
|
|
| 33 |
E |
836 |
803 |
4.68 |
|
|
|
| 33 |
E |
836 |
803 |
4.68 |
|
|
|
| 34 |
E |
590 |
567 |
0.00 |
|
|
|
| 34 |
E |
590 |
567 |
0.00 |
|
|
|
| 35 |
E |
428 |
411 |
0.00 |
|
|
|
| 35 |
E |
428 |
411 |
0.00 |
|
|
|
| 36 |
E |
340 |
327 |
0.00 |
|
|
|
| 36 |
E |
340 |
327 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40486.8 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 38879.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G*
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.290 |
| N2 |
0.000 |
0.000 |
-1.290 |
| C3 |
-0.000 |
1.384 |
0.783 |
| C4 |
1.198 |
-0.691 |
0.783 |
| C5 |
-1.198 |
-0.692 |
0.783 |
| C6 |
0.000 |
1.384 |
-0.783 |
| C7 |
-1.198 |
-0.691 |
-0.783 |
| C8 |
1.198 |
-0.692 |
-0.783 |
| H9 |
0.883 |
1.894 |
1.186 |
| H10 |
-0.885 |
1.892 |
1.185 |
| H11 |
1.198 |
-1.711 |
1.186 |
| H12 |
2.081 |
-0.180 |
1.185 |
| H13 |
-2.081 |
-0.182 |
1.186 |
| H14 |
-1.196 |
-1.712 |
1.185 |
| H15 |
-0.883 |
1.894 |
-1.186 |
| H16 |
0.885 |
1.892 |
-1.185 |
| H17 |
-1.198 |
-1.711 |
-1.186 |
| H18 |
-2.081 |
-0.180 |
-1.185 |
| H19 |
2.081 |
-0.182 |
-1.186 |
| H20 |
1.196 |
-1.712 |
-1.185 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5802 | 1.4737 | 1.4737 | 1.4737 | 2.4921 | 2.4921 | 2.4921 | 2.0918 | 2.0915 | 2.0918 | 2.0915 | 2.0918 | 2.0915 | 3.2396 | 3.2387 | 3.2396 | 3.2387 | 3.2396 | 3.2387 |
N2 | 2.5802 | | 2.4921 | 2.4921 | 2.4921 | 1.4737 | 1.4737 | 1.4737 | 3.2396 | 3.2387 | 3.2396 | 3.2387 | 3.2396 | 3.2387 | 2.0918 | 2.0915 | 2.0918 | 2.0915 | 2.0918 | 2.0915 | C3 | 1.4737 | 2.4921 | | 2.3964 | 2.3964 | 1.5653 | 2.8621 | 2.8627 | 1.0967 | 1.0967 | 3.3434 | 2.6342 | 2.6352 | 3.3432 | 2.2167 | 2.2167 | 3.8586 | 3.2628 | 3.2649 | 3.8586 | C4 | 1.4737 | 2.4921 | 2.3964 | | 2.3964 | 2.8621 | 2.8627 | 1.5653 | 2.6352 | 3.3432 | 1.0967 | 1.0967 | 3.3434 | 2.6342 | 3.8586 | 3.2628 | 3.2649 | 3.8586 | 2.2167 | 2.2167 | C5 | 1.4737 | 2.4921 | 2.3964 | 2.3964 | | 2.8627 | 1.5653 | 2.8621 | 3.3434 | 2.6342 | 2.6352 | 3.3432 | 1.0967 | 1.0967 | 3.2649 | 3.8586 | 2.2167 | 2.2167 | 3.8586 | 3.2628 | C6 | 2.4921 | 1.4737 | 1.5653 | 2.8621 | 2.8627 | | 2.3964 | 2.3964 | 2.2167 | 2.2167 | 3.8586 | 3.2628 | 3.2649 | 3.8586 | 1.0967 | 1.0967 | 3.3434 | 2.6342 | 2.6352 | 3.3432 | C7 | 2.4921 | 1.4737 | 2.8621 | 2.8627 | 1.5653 | 2.3964 | | 2.3964 | 3.8586 | 3.2628 | 3.2649 | 3.8586 | 2.2167 | 2.2167 | 2.6352 | 3.3432 | 1.0967 | 1.0967 | 3.3434 | 2.6342 | C8 | 2.4921 | 1.4737 | 2.8627 | 1.5653 | 2.8621 | 2.3964 | 2.3964 | | 3.2649 | 3.8586 | 2.2167 | 2.2167 | 3.8586 | 3.2628 | 3.3434 | 2.6342 | 2.6352 | 3.3432 | 1.0967 | 1.0967 | H9 | 2.0918 | 3.2396 | 1.0967 | 2.6352 | 3.3434 | 2.2167 | 3.8586 | 3.2649 | | 1.7677 | 3.6187 | 2.3947 | 3.6187 | 4.1624 | 2.9569 | 2.3708 | 4.7908 | 4.3250 | 3.3718 | 4.3268 | H10 | 2.0915 | 3.2387 | 1.0967 | 3.3432 | 2.6342 | 2.2167 | 3.2628 | 3.8586 | 1.7677 | | 4.1624 | 3.6181 | 2.3947 | 3.6181 | 2.3708 | 2.9577 | 4.3250 | 3.3677 | 4.3268 | 4.7897 | H11 | 2.0918 | 3.2396 | 3.3434 | 1.0967 | 2.6352 | 3.8586 | 3.2649 | 2.2167 | 3.6187 | 4.1624 | | 1.7677 | 3.6187 | 2.3947 | 4.7908 | 4.3250 | 3.3718 | 4.3268 | 2.9569 | 2.3708 | H12 | 2.0915 | 3.2387 | 2.6342 | 1.0967 | 3.3432 | 3.2628 | 3.8586 | 2.2167 | 2.3947 | 3.6181 | 1.7677 | | 4.1624 | 3.6181 | 4.3250 | 3.3677 | 4.3268 | 4.7897 | 2.3708 | 2.9577 | H13 | 2.0918 | 3.2396 | 2.6352 | 3.3434 | 1.0967 | 3.2649 | 2.2167 | 3.8586 | 3.6187 | 2.3947 | 3.6187 | 4.1624 | | 1.7677 | 3.3718 | 4.3268 | 2.9569 | 2.3708 | 4.7908 | 4.3250 | H14 | 2.0915 | 3.2387 | 3.3432 | 2.6342 | 1.0967 | 3.8586 | 2.2167 | 3.2628 | 4.1624 | 3.6181 | 2.3947 | 3.6181 | 1.7677 | | 4.3268 | 4.7897 | 2.3708 | 2.9577 | 4.3250 | 3.3677 | H15 | 3.2396 | 2.0918 | 2.2167 | 3.8586 | 3.2649 | 1.0967 | 2.6352 | 3.3434 | 2.9569 | 2.3708 | 4.7908 | 4.3250 | 3.3718 | 4.3268 | | 1.7677 | 3.6187 | 2.3947 | 3.6187 | 4.1624 | H16 | 3.2387 | 2.0915 | 2.2167 | 3.2628 | 3.8586 | 1.0967 | 3.3432 | 2.6342 | 2.3708 | 2.9577 | 4.3250 | 3.3677 | 4.3268 | 4.7897 | 1.7677 | | 4.1624 | 3.6181 | 2.3947 | 3.6181 | H17 | 3.2396 | 2.0918 | 3.8586 | 3.2649 | 2.2167 | 3.3434 | 1.0967 | 2.6352 | 4.7908 | 4.3250 | 3.3718 | 4.3268 | 2.9569 | 2.3708 | 3.6187 | 4.1624 | | 1.7677 | 3.6187 | 2.3947 | H18 | 3.2387 | 2.0915 | 3.2628 | 3.8586 | 2.2167 | 2.6342 | 1.0967 | 3.3432 | 4.3250 | 3.3677 | 4.3268 | 4.7897 | 2.3708 | 2.9577 | 2.3947 | 3.6181 | 1.7677 | | 4.1624 | 3.6181 | H19 | 3.2396 | 2.0918 | 3.2649 | 2.2167 | 3.8586 | 2.6352 | 3.3434 | 1.0967 | 3.3718 | 4.3268 | 2.9569 | 2.3708 | 4.7908 | 4.3250 | 3.6187 | 2.3947 | 3.6187 | 4.1624 | | 1.7677 | H20 | 3.2387 | 2.0915 | 3.8586 | 2.2167 | 3.2628 | 3.3432 | 2.6342 | 1.0967 | 4.3268 | 4.7897 | 2.3708 | 2.9577 | 4.3250 | 3.3677 | 4.1624 | 3.6181 | 2.3947 | 3.6181 | 1.7677 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
110.141 |
|
N1 |
C3 |
H9 |
108.043 |
| N1 |
C3 |
H10 |
108.019 |
|
N1 |
C4 |
C8 |
110.141 |
| N1 |
C4 |
H11 |
108.043 |
|
N1 |
C4 |
H12 |
108.019 |
| N1 |
C5 |
C7 |
110.141 |
|
N1 |
C5 |
H13 |
108.043 |
| N1 |
C5 |
H14 |
108.019 |
|
N2 |
C6 |
C3 |
110.141 |
| N2 |
C6 |
H15 |
108.043 |
|
N2 |
C6 |
H16 |
108.019 |
| N2 |
C7 |
C5 |
110.141 |
|
N2 |
C7 |
H17 |
108.043 |
| N2 |
C7 |
H18 |
108.019 |
|
N2 |
C8 |
C4 |
110.141 |
| N2 |
C8 |
H19 |
108.043 |
|
N2 |
C8 |
H20 |
108.019 |
| C3 |
N1 |
C4 |
108.793 |
|
C3 |
N1 |
C5 |
108.793 |
| C3 |
C6 |
H15 |
111.548 |
|
C3 |
C6 |
H16 |
111.543 |
| C4 |
N1 |
C5 |
108.793 |
|
C4 |
C8 |
H19 |
111.548 |
| C4 |
C8 |
H20 |
111.543 |
|
C5 |
C6 |
H15 |
101.596 |
| C5 |
C6 |
H16 |
150.984 |
|
C6 |
N2 |
C7 |
108.793 |
| C6 |
N2 |
C8 |
108.793 |
|
C6 |
C3 |
H9 |
111.548 |
| C6 |
C3 |
H10 |
111.543 |
|
C7 |
N2 |
C8 |
108.793 |
| C7 |
C5 |
H13 |
111.548 |
|
C7 |
C5 |
H14 |
111.543 |
| C8 |
C4 |
H11 |
111.548 |
|
C8 |
C4 |
H12 |
111.543 |
| H9 |
C3 |
H10 |
107.395 |
|
H11 |
C4 |
H12 |
107.396 |
| H13 |
C5 |
H14 |
107.395 |
|
H15 |
C6 |
H16 |
107.395 |
| H17 |
C7 |
H18 |
107.396 |
|
H19 |
C8 |
H20 |
107.395 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.367 |
|
|
|
| 2 |
N |
-0.367 |
|
|
|
| 3 |
C |
-0.157 |
|
|
|
| 4 |
C |
-0.157 |
|
|
|
| 5 |
C |
-0.157 |
|
|
|
| 6 |
C |
-0.157 |
|
|
|
| 7 |
C |
-0.157 |
|
|
|
| 8 |
C |
-0.157 |
|
|
|
| 9 |
H |
0.140 |
|
|
|
| 10 |
H |
0.140 |
|
|
|
| 11 |
H |
0.140 |
|
|
|
| 12 |
H |
0.140 |
|
|
|
| 13 |
H |
0.140 |
|
|
|
| 14 |
H |
0.140 |
|
|
|
| 15 |
H |
0.140 |
|
|
|
| 16 |
H |
0.140 |
|
|
|
| 17 |
H |
0.140 |
|
|
|
| 18 |
H |
0.140 |
|
|
|
| 19 |
H |
0.140 |
|
|
|
| 20 |
H |
0.140 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.725 |
0.000 |
0.000 |
| y |
0.000 |
-46.725 |
0.000 |
| z |
0.000 |
0.000 |
-57.372 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.323 |
0.000 |
0.000 |
| y |
0.000 |
5.323 |
0.000 |
| z |
0.000 |
0.000 |
-10.647 |
|
| Polar |
|---|
| 3z2-r2 | -21.294 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.945 |
0.000 |
0.000 |
| y |
0.000 |
10.945 |
0.000 |
| z |
0.000 |
0.000 |
9.352 |
<r2> (average value of r
2) Å
2
| <r2> |
214.901 |
| (<r2>)1/2 |
14.659 |