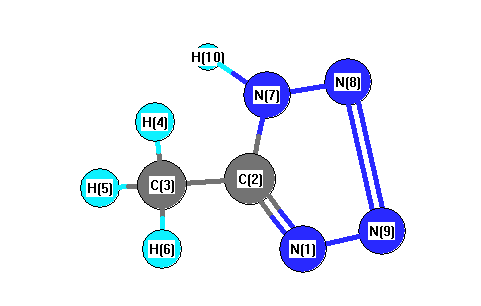
Vibrational Frequencies calculated at B3LYP/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3649 |
3504 |
66.31 |
|
|
|
| 2 |
A |
3169 |
3043 |
1.77 |
|
|
|
| 3 |
A |
3062 |
2940 |
12.21 |
|
|
|
| 4 |
A |
1597 |
1534 |
44.63 |
|
|
|
| 5 |
A |
1523 |
1462 |
0.86 |
|
|
|
| 6 |
A |
1445 |
1388 |
0.51 |
|
|
|
| 7 |
A |
1428 |
1371 |
27.77 |
|
|
|
| 8 |
A |
1372 |
1317 |
3.78 |
|
|
|
| 9 |
A |
1282 |
1231 |
20.08 |
|
|
|
| 10 |
A |
1113 |
1069 |
3.49 |
|
|
|
| 11 |
A |
1084 |
1041 |
12.29 |
|
|
|
| 12 |
A |
1059 |
1017 |
22.79 |
|
|
|
| 13 |
A |
1015 |
975 |
0.10 |
|
|
|
| 14 |
A |
1000 |
960 |
1.65 |
|
|
|
| 15 |
A |
687 |
660 |
3.25 |
|
|
|
| 16 |
A |
337 |
324 |
3.91 |
|
|
|
| 17 |
A |
3117 |
2994 |
9.58 |
|
|
|
| 18 |
A |
1514 |
1454 |
9.89 |
|
|
|
| 19 |
A |
1082 |
1039 |
2.32 |
|
|
|
| 20 |
A |
735 |
706 |
5.43 |
|
|
|
| 21 |
A |
704 |
676 |
16.01 |
|
|
|
| 22 |
A |
562 |
540 |
78.64 |
|
|
|
| 23 |
A |
268 |
258 |
0.06 |
|
|
|
| 24 |
A |
83 |
80 |
0.46 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 16442.6 cm
-1
Scaled (by 0.9603) Zero Point Vibrational Energy (zpe) 15789.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.357 |
-0.416 |
|
-0.440 |
| 2 |
C |
0.556 |
0.570 |
|
0.624 |
| 3 |
C |
-0.524 |
-0.493 |
|
-0.625 |
| 4 |
H |
0.182 |
0.128 |
|
0.161 |
| 5 |
H |
0.182 |
0.128 |
|
0.161 |
| 6 |
H |
0.209 |
0.158 |
|
0.196 |
| 7 |
N |
-0.472 |
-0.000 |
|
0.000 |
| 8 |
N |
-0.043 |
-0.251 |
|
-0.257 |
| 9 |
N |
-0.089 |
-0.050 |
|
-0.046 |
| 10 |
H |
0.357 |
0.224 |
|
0.225 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
4.135 |
3.979 |
0.000 |
5.738 |
| CHELPG |
3.951 |
3.748 |
0.000 |
5.446 |
| AIM |
|
|
|
|
| ESP |
4.025 |
3.823 |
0.000 |
5.551 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-33.562 |
-0.384 |
0.000 |
| y |
-0.384 |
-36.447 |
0.000 |
| z |
0.000 |
0.000 |
-34.164 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.744 |
-0.384 |
0.000 |
| y |
-0.384 |
-2.584 |
0.000 |
| z |
0.000 |
0.000 |
0.841 |
|
| Polar |
|---|
| 3z2-r2 | 1.681 |
|---|
| x2-y2 | 2.885 |
|---|
| xy | -0.384 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.687 |
0.286 |
0.000 |
| y |
0.286 |
8.010 |
0.000 |
| z |
0.000 |
0.000 |
3.720 |
<r2> (average value of r
2) Å
2
| <r2> |
128.998 |
| (<r2>)1/2 |
11.358 |