Vibrational Frequencies calculated at B3LYP/6-311+G(3df,2p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3155 |
3051 |
7.34 |
|
|
|
| 2 |
A' |
3120 |
3017 |
19.24 |
|
|
|
| 3 |
A' |
3108 |
3006 |
10.53 |
|
|
|
| 4 |
A' |
3030 |
2930 |
13.18 |
|
|
|
| 5 |
A' |
3016 |
2917 |
30.60 |
|
|
|
| 6 |
A' |
1722 |
1665 |
12.50 |
|
|
|
| 7 |
A' |
1497 |
1448 |
8.32 |
|
|
|
| 8 |
A' |
1487 |
1438 |
5.19 |
|
|
|
| 9 |
A' |
1425 |
1378 |
5.20 |
|
|
|
| 10 |
A' |
1414 |
1367 |
0.90 |
|
|
|
| 11 |
A' |
1358 |
1313 |
0.96 |
|
|
|
| 12 |
A' |
1152 |
1114 |
33.31 |
|
|
|
| 13 |
A' |
1081 |
1046 |
29.33 |
|
|
|
| 14 |
A' |
1007 |
973 |
10.45 |
|
|
|
| 15 |
A' |
912 |
882 |
20.63 |
|
|
|
| 16 |
A' |
674 |
652 |
20.90 |
|
|
|
| 17 |
A' |
438 |
423 |
8.53 |
|
|
|
| 18 |
A' |
349 |
338 |
0.40 |
|
|
|
| 19 |
A' |
271 |
262 |
0.38 |
|
|
|
| 20 |
A" |
3077 |
2975 |
8.91 |
|
|
|
| 21 |
A" |
3054 |
2953 |
15.25 |
|
|
|
| 22 |
A" |
1485 |
1436 |
10.38 |
|
|
|
| 23 |
A" |
1477 |
1428 |
5.10 |
|
|
|
| 24 |
A" |
1066 |
1031 |
0.34 |
|
|
|
| 25 |
A" |
1064 |
1029 |
1.56 |
|
|
|
| 26 |
A" |
861 |
832 |
15.53 |
|
|
|
| 27 |
A" |
451 |
436 |
3.68 |
|
|
|
| 28 |
A" |
232 |
224 |
0.54 |
|
|
|
| 29 |
A" |
171 |
166 |
0.28 |
|
|
|
| 30 |
A" |
129 |
125 |
0.54 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21640.9 cm
-1
Scaled (by 0.967) Zero Point Vibrational Energy (zpe) 20926.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
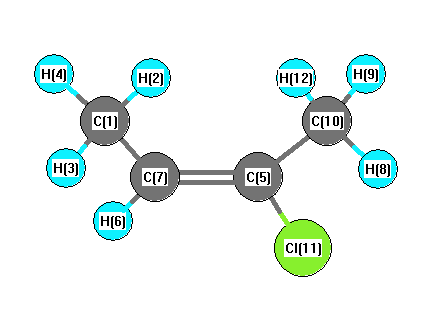
Charges from optimized geometry at B3LYP/6-311+G(3df,2p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.417 |
|
|
|
| 2 |
H |
0.139 |
|
|
|
| 3 |
H |
0.154 |
|
|
|
| 4 |
H |
0.154 |
|
|
|
| 5 |
C |
0.478 |
|
|
|
| 6 |
H |
0.141 |
|
|
|
| 7 |
C |
-0.293 |
|
|
|
| 8 |
H |
0.160 |
|
|
|
| 9 |
H |
0.160 |
|
|
|
| 10 |
C |
-0.586 |
|
|
|
| 11 |
Cl |
-0.230 |
|
|
|
| 12 |
H |
0.141 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-2.141 |
0.160 |
0.000 |
2.147 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.009 |
1.431 |
0.000 |
| y |
1.431 |
10.402 |
0.000 |
| z |
0.000 |
0.000 |
7.191 |
<r2> (average value of r
2) Å
2
| <r2> |
184.185 |
| (<r2>)1/2 |
13.571 |