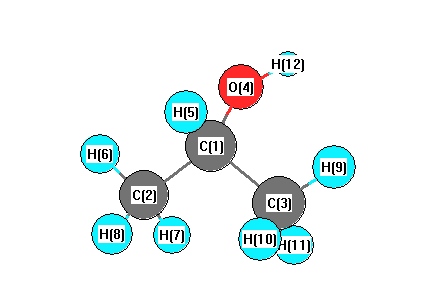
Vibrational Frequencies calculated at B3LYP/CEP-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3788 |
3658 |
7.46 |
|
|
|
| 2 |
A |
3127 |
3020 |
47.08 |
|
|
|
| 3 |
A |
3123 |
3016 |
84.58 |
|
|
|
| 4 |
A |
3112 |
3006 |
17.72 |
|
|
|
| 5 |
A |
3094 |
2987 |
53.34 |
|
|
|
| 6 |
A |
3049 |
2945 |
17.87 |
|
|
|
| 7 |
A |
3031 |
2927 |
32.46 |
|
|
|
| 8 |
A |
2994 |
2891 |
76.50 |
|
|
|
| 9 |
A |
1508 |
1457 |
6.99 |
|
|
|
| 10 |
A |
1496 |
1444 |
4.33 |
|
|
|
| 11 |
A |
1487 |
1436 |
1.37 |
|
|
|
| 12 |
A |
1481 |
1431 |
1.10 |
|
|
|
| 13 |
A |
1430 |
1381 |
23.93 |
|
|
|
| 14 |
A |
1411 |
1363 |
19.39 |
|
|
|
| 15 |
A |
1380 |
1333 |
0.60 |
|
|
|
| 16 |
A |
1358 |
1311 |
21.85 |
|
|
|
| 17 |
A |
1274 |
1231 |
58.97 |
|
|
|
| 18 |
A |
1176 |
1136 |
34.84 |
|
|
|
| 19 |
A |
1145 |
1106 |
33.11 |
|
|
|
| 20 |
A |
1080 |
1043 |
20.07 |
|
|
|
| 21 |
A |
959 |
926 |
34.30 |
|
|
|
| 22 |
A |
939 |
907 |
1.83 |
|
|
|
| 23 |
A |
914 |
883 |
1.25 |
|
|
|
| 24 |
A |
813 |
786 |
3.76 |
|
|
|
| 25 |
A |
461 |
445 |
7.53 |
|
|
|
| 26 |
A |
402 |
388 |
10.33 |
|
|
|
| 27 |
A |
352 |
340 |
3.69 |
|
|
|
| 28 |
A |
308 |
297 |
125.18 |
|
|
|
| 29 |
A |
259 |
250 |
2.82 |
|
|
|
| 30 |
A |
219 |
211 |
3.98 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23583.9 cm
-1
Scaled (by 0.9657) Zero Point Vibrational Energy (zpe) 22775.0 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.018 |
|
|
|
| 2 |
C |
-0.487 |
|
|
|
| 3 |
C |
-0.440 |
|
|
|
| 4 |
O |
-0.483 |
|
|
|
| 5 |
H |
0.127 |
|
|
|
| 6 |
H |
0.154 |
|
|
|
| 7 |
H |
0.157 |
|
|
|
| 8 |
H |
0.140 |
|
|
|
| 9 |
H |
0.177 |
|
|
|
| 10 |
H |
0.134 |
|
|
|
| 11 |
H |
0.144 |
|
|
|
| 12 |
H |
0.359 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.166 |
-0.939 |
0.946 |
1.771 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.706 |
-2.749 |
-0.331 |
| y |
-2.749 |
-26.949 |
1.100 |
| z |
-0.331 |
1.100 |
-26.163 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.850 |
-2.749 |
-0.331 |
| y |
-2.749 |
-1.514 |
1.100 |
| z |
-0.331 |
1.100 |
-0.336 |
|
| Polar |
|---|
| 3z2-r2 | -0.672 |
|---|
| x2-y2 | 2.243 |
|---|
| xy | -2.749 |
|---|
| xz | -0.331 |
|---|
| yz | 1.100 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.008 |
-0.163 |
-0.074 |
| y |
-0.163 |
5.567 |
-0.033 |
| z |
-0.074 |
-0.033 |
4.838 |
<r2> (average value of r
2) Å
2
| <r2> |
78.023 |
| (<r2>)1/2 |
8.833 |