Jump to
S1C2
Energy calculated at B3LYP/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -231.350010 |
| Energy at 298.15K | |
| HF Energy | -231.350010 |
| Nuclear repulsion energy | 369.362603 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
2006 |
1954 |
0.94 |
|
|
|
| 2 |
A |
1924 |
1875 |
957.25 |
|
|
|
| 3 |
A |
1916 |
1867 |
0.12 |
|
|
|
| 4 |
A |
1912 |
1863 |
1491.48 |
|
|
|
| 5 |
A |
1912 |
1863 |
1491.65 |
|
|
|
| 6 |
A |
654 |
638 |
140.89 |
|
|
|
| 7 |
A |
631 |
615 |
107.84 |
|
|
|
| 8 |
A |
631 |
615 |
107.82 |
|
|
|
| 9 |
A |
549 |
535 |
0.00 |
|
|
|
| 10 |
A |
516 |
503 |
21.92 |
|
|
|
| 11 |
A |
516 |
503 |
21.96 |
|
|
|
| 12 |
A |
486 |
473 |
3.10 |
|
|
|
| 13 |
A |
477 |
465 |
4.78 |
|
|
|
| 14 |
A |
477 |
465 |
4.72 |
|
|
|
| 15 |
A |
432 |
421 |
0.00 |
|
|
|
| 16 |
A |
412 |
402 |
0.04 |
|
|
|
| 17 |
A |
355 |
346 |
0.01 |
|
|
|
| 18 |
A |
355 |
346 |
3.34 |
|
|
|
| 19 |
A |
355 |
346 |
3.35 |
|
|
|
| 20 |
A |
322 |
314 |
0.00 |
|
|
|
| 21 |
A |
109 |
107 |
1.30 |
|
|
|
| 22 |
A |
109 |
107 |
1.30 |
|
|
|
| 23 |
A |
107 |
105 |
1.03 |
|
|
|
| 24 |
A |
106 |
103 |
0.00 |
|
|
|
| 25 |
A |
84 |
82 |
0.00 |
|
|
|
| 26 |
A |
84 |
82 |
0.00 |
|
|
|
| 27 |
A |
43i |
42i |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8697.2 cm
-1
Scaled (by 0.9745) Zero Point Vibrational Energy (zpe) 8475.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/CEP-121G
Point Group is C1
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-121G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Fe |
-1.032 |
|
|
|
| 2 |
C |
0.200 |
|
|
|
| 3 |
C |
0.132 |
|
|
|
| 4 |
C |
0.131 |
|
|
|
| 5 |
C |
0.132 |
|
|
|
| 6 |
C |
0.131 |
|
|
|
| 7 |
O |
0.055 |
|
|
|
| 8 |
O |
0.063 |
|
|
|
| 9 |
O |
0.063 |
|
|
|
| 10 |
O |
0.063 |
|
|
|
| 11 |
O |
0.063 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.001 |
0.001 |
0.067 |
0.067 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-74.640 |
-0.001 |
-0.003 |
| y |
-0.001 |
-74.651 |
0.002 |
| z |
-0.003 |
0.002 |
-73.370 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.629 |
-0.001 |
-0.003 |
| y |
-0.001 |
-0.646 |
0.002 |
| z |
-0.003 |
0.002 |
1.275 |
|
| Polar |
|---|
| 3z2-r2 | 2.550 |
|---|
| x2-y2 | 0.012 |
|---|
| xy | -0.001 |
|---|
| xz | -0.003 |
|---|
| yz | 0.002 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
15.859 |
0.000 |
0.002 |
| y |
0.000 |
15.857 |
-0.002 |
| z |
0.002 |
-0.002 |
12.434 |
<r2> (average value of r
2) Å
2
| <r2> |
383.333 |
| (<r2>)1/2 |
19.579 |
Jump to
S1C1
Energy calculated at B3LYP/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -231.352717 |
| Energy at 298.15K | -231.351203 |
| HF Energy | -231.352717 |
| Nuclear repulsion energy | 369.243654 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1' |
2009 |
1958 |
0.00 |
|
|
|
| 2 |
A1' |
1930 |
1881 |
0.00 |
|
|
|
| 3 |
A1' |
432 |
421 |
0.00 |
|
|
|
| 4 |
A1' |
411 |
400 |
0.00 |
|
|
|
| 5 |
A2' |
353 |
344 |
0.00 |
|
|
|
| 6 |
A2" |
1934 |
1885 |
1446.69 |
|
|
|
| 7 |
A2" |
603 |
588 |
129.89 |
|
|
|
| 8 |
A2" |
471 |
459 |
3.91 |
|
|
|
| 9 |
A2" |
109 |
106 |
1.50 |
|
|
|
| 10 |
E' |
1903 |
1854 |
1260.20 |
|
|
|
| 10 |
E' |
1903 |
1854 |
1258.54 |
|
|
|
| 11 |
E' |
644 |
628 |
142.98 |
|
|
|
| 11 |
E' |
644 |
628 |
142.22 |
|
|
|
| 12 |
E' |
482 |
469 |
1.33 |
|
|
|
| 12 |
E' |
482 |
469 |
1.34 |
|
|
|
| 13 |
E' |
425 |
414 |
11.74 |
|
|
|
| 13 |
E' |
425 |
414 |
11.61 |
|
|
|
| 14 |
E' |
103 |
101 |
0.89 |
|
|
|
| 14 |
E' |
103 |
101 |
0.90 |
|
|
|
| 15 |
E' |
48 |
47 |
0.00 |
|
|
|
| 15 |
E' |
48 |
47 |
0.00 |
|
|
|
| 16 |
E" |
548 |
534 |
0.00 |
|
|
|
| 16 |
E" |
548 |
534 |
0.00 |
|
|
|
| 17 |
E" |
350 |
341 |
0.00 |
|
|
|
| 17 |
E" |
350 |
341 |
0.00 |
|
|
|
| 18 |
E" |
98 |
96 |
0.00 |
|
|
|
| 18 |
E" |
98 |
96 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 8726.9 cm
-1
Scaled (by 0.9745) Zero Point Vibrational Energy (zpe) 8504.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/CEP-121G
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| Fe1 |
0.000 |
0.000 |
0.000 |
| C2 |
0.000 |
0.000 |
1.829 |
| C3 |
0.000 |
1.817 |
0.000 |
| C4 |
1.573 |
-0.908 |
0.000 |
| C5 |
-1.573 |
-0.908 |
0.000 |
| C6 |
0.000 |
0.000 |
-1.829 |
| O7 |
0.000 |
0.000 |
3.007 |
| O8 |
0.000 |
2.999 |
0.000 |
| O9 |
2.598 |
-1.500 |
0.000 |
| O10 |
-2.598 |
-1.500 |
0.000 |
| O11 |
0.000 |
0.000 |
-3.007 |
Atom - Atom Distances (Å)
| |
Fe1 |
C2 |
C3 |
C4 |
C5 |
C6 |
O7 |
O8 |
O9 |
O10 |
O11 |
| Fe1 | | 1.8288 | 1.8165 | 1.8165 | 1.8165 | 1.8288 | 3.0068 | 2.9995 | 2.9995 | 2.9995 | 3.0068 |
C2 | 1.8288 | | 2.5777 | 2.5777 | 2.5777 | 3.6576 | 1.1780 | 3.5130 | 3.5130 | 3.5130 | 4.8356 | C3 | 1.8165 | 2.5777 | | 3.1464 | 3.1464 | 2.5777 | 3.5129 | 1.1829 | 4.2125 | 4.2125 | 3.5129 | C4 | 1.8165 | 2.5777 | 3.1464 | | 3.1464 | 2.5777 | 3.5129 | 4.2125 | 1.1829 | 4.2125 | 3.5129 | C5 | 1.8165 | 2.5777 | 3.1464 | 3.1464 | | 2.5777 | 3.5129 | 4.2125 | 4.2125 | 1.1829 | 3.5129 | C6 | 1.8288 | 3.6576 | 2.5777 | 2.5777 | 2.5777 | | 4.8356 | 3.5130 | 3.5130 | 3.5130 | 1.1780 | O7 | 3.0068 | 1.1780 | 3.5129 | 3.5129 | 3.5129 | 4.8356 | | 4.2471 | 4.2471 | 4.2471 | 6.0136 | O8 | 2.9995 | 3.5130 | 1.1829 | 4.2125 | 4.2125 | 3.5130 | 4.2471 | | 5.1952 | 5.1952 | 4.2471 | O9 | 2.9995 | 3.5130 | 4.2125 | 1.1829 | 4.2125 | 3.5130 | 4.2471 | 5.1952 | | 5.1952 | 4.2471 | O10 | 2.9995 | 3.5130 | 4.2125 | 4.2125 | 1.1829 | 3.5130 | 4.2471 | 5.1952 | 5.1952 | | 4.2471 | O11 | 3.0068 | 4.8356 | 3.5129 | 3.5129 | 3.5129 | 1.1780 | 6.0136 | 4.2471 | 4.2471 | 4.2471 | |
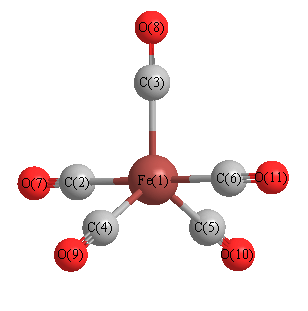 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Fe1 |
C2 |
O7 |
180.000 |
|
Fe1 |
C3 |
O8 |
180.000 |
| Fe1 |
C4 |
O9 |
180.000 |
|
Fe1 |
C5 |
O10 |
180.000 |
| Fe1 |
C6 |
O11 |
180.000 |
|
C2 |
Fe1 |
C3 |
90.000 |
| C2 |
Fe1 |
C4 |
90.000 |
|
C2 |
Fe1 |
C5 |
90.000 |
| C2 |
Fe1 |
C6 |
180.000 |
|
C3 |
Fe1 |
C4 |
120.000 |
| C3 |
Fe1 |
C5 |
120.000 |
|
C3 |
Fe1 |
C6 |
90.000 |
| C4 |
Fe1 |
C5 |
120.000 |
|
C4 |
Fe1 |
C6 |
90.000 |
| C5 |
Fe1 |
C6 |
90.000 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-121G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
Fe |
-1.084 |
|
|
|
| 2 |
C |
0.160 |
|
|
|
| 3 |
C |
0.156 |
|
|
|
| 4 |
C |
0.156 |
|
|
|
| 5 |
C |
0.156 |
|
|
|
| 6 |
C |
0.160 |
|
|
|
| 7 |
O |
0.076 |
|
|
|
| 8 |
O |
0.049 |
|
|
|
| 9 |
O |
0.049 |
|
|
|
| 10 |
O |
0.049 |
|
|
|
| 11 |
O |
0.076 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-74.706 |
0.000 |
0.000 |
| y |
0.000 |
-74.706 |
0.000 |
| z |
0.000 |
0.000 |
-73.149 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.779 |
0.000 |
0.000 |
| y |
0.000 |
-0.779 |
0.000 |
| z |
0.000 |
0.000 |
1.557 |
|
| Polar |
|---|
| 3z2-r2 | 3.114 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
14.107 |
0.000 |
0.000 |
| y |
0.000 |
14.086 |
0.000 |
| z |
0.000 |
0.000 |
15.795 |
<r2> (average value of r
2) Å
2
| <r2> |
383.124 |
| (<r2>)1/2 |
19.574 |