Jump to
S1C2
Energy calculated at B3LYP/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -62.838461 |
| Energy at 298.15K | |
| HF Energy | -62.838461 |
| Nuclear repulsion energy | 128.361822 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3081 |
3003 |
51.77 |
|
|
|
| 2 |
A' |
3017 |
2940 |
57.04 |
|
|
|
| 3 |
A' |
3011 |
2934 |
8.16 |
|
|
|
| 4 |
A' |
2986 |
2910 |
35.96 |
|
|
|
| 5 |
A' |
1559 |
1519 |
160.53 |
|
|
|
| 6 |
A' |
1536 |
1497 |
26.44 |
|
|
|
| 7 |
A' |
1522 |
1483 |
1.57 |
|
|
|
| 8 |
A' |
1516 |
1477 |
16.88 |
|
|
|
| 9 |
A' |
1438 |
1402 |
10.34 |
|
|
|
| 10 |
A' |
1404 |
1368 |
22.98 |
|
|
|
| 11 |
A' |
1340 |
1306 |
11.58 |
|
|
|
| 12 |
A' |
1150 |
1120 |
0.51 |
|
|
|
| 13 |
A' |
1048 |
1021 |
2.82 |
|
|
|
| 14 |
A' |
986 |
961 |
33.58 |
|
|
|
| 15 |
A' |
921 |
897 |
72.77 |
|
|
|
| 16 |
A' |
826 |
805 |
425.91 |
|
|
|
| 17 |
A' |
617 |
601 |
7.46 |
|
|
|
| 18 |
A' |
364 |
355 |
0.50 |
|
|
|
| 19 |
A' |
337 |
329 |
0.46 |
|
|
|
| 20 |
A' |
145 |
141 |
0.52 |
|
|
|
| 21 |
A" |
3095 |
3016 |
103.60 |
|
|
|
| 22 |
A" |
3070 |
2992 |
10.91 |
|
|
|
| 23 |
A" |
3050 |
2972 |
3.29 |
|
|
|
| 24 |
A" |
1525 |
1486 |
9.69 |
|
|
|
| 25 |
A" |
1316 |
1282 |
0.00 |
|
|
|
| 26 |
A" |
1257 |
1225 |
0.20 |
|
|
|
| 27 |
A" |
1174 |
1144 |
1.09 |
|
|
|
| 28 |
A" |
899 |
876 |
1.25 |
|
|
|
| 29 |
A" |
771 |
751 |
3.09 |
|
|
|
| 30 |
A" |
229 |
224 |
0.00 |
|
|
|
| 31 |
A" |
197 |
192 |
1.34 |
|
|
|
| 32 |
A" |
86 |
84 |
2.38 |
|
|
|
| 33 |
A" |
47i |
46i |
0.25 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22712.6 cm
-1
Scaled (by 0.9745) Zero Point Vibrational Energy (zpe) 22133.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/CEP-121G
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-1.945 |
2.281 |
0.000 |
| C2 |
-1.530 |
0.786 |
0.000 |
| C3 |
0.000 |
0.626 |
0.000 |
| O4 |
0.279 |
-0.843 |
0.000 |
| N5 |
1.726 |
-1.092 |
0.000 |
| O6 |
1.959 |
-2.304 |
0.000 |
| H7 |
-3.038 |
2.375 |
0.000 |
| H8 |
-1.564 |
2.803 |
0.889 |
| H9 |
-1.564 |
2.803 |
-0.889 |
| H10 |
-1.941 |
0.278 |
-0.883 |
| H11 |
-1.941 |
0.278 |
0.883 |
| H12 |
0.456 |
1.066 |
0.896 |
| H13 |
0.456 |
1.066 |
-0.896 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5516 | 2.5537 | 3.8347 | 4.9849 | 6.0216 | 1.0971 | 1.0990 | 1.0990 | 2.1884 | 2.1884 | 2.8362 | 2.8362 |
C2 | 1.5516 | | 1.5385 | 2.4344 | 3.7585 | 4.6606 | 2.1905 | 2.2051 | 2.2051 | 1.0980 | 1.0980 | 2.1970 | 2.1970 | C3 | 2.5537 | 1.5385 | | 1.4952 | 2.4350 | 3.5244 | 3.5053 | 2.8246 | 2.8246 | 2.1607 | 2.1607 | 1.0976 | 1.0976 | O4 | 3.8347 | 2.4344 | 1.4952 | | 1.4678 | 2.2263 | 4.6214 | 4.1815 | 4.1815 | 2.6396 | 2.6396 | 2.1162 | 2.1162 | N5 | 4.9849 | 3.7585 | 2.4350 | 1.4678 | | 1.2338 | 5.8915 | 5.1757 | 5.1757 | 4.0130 | 4.0130 | 2.6593 | 2.6593 | O6 | 6.0216 | 4.6606 | 3.5244 | 2.2263 | 1.2338 | | 6.8454 | 6.2680 | 6.2680 | 4.7604 | 4.7604 | 3.7969 | 3.7969 | H7 | 1.0971 | 2.1905 | 3.5053 | 4.6214 | 5.8915 | 6.8454 | | 1.7735 | 1.7735 | 2.5252 | 2.5252 | 3.8372 | 3.8372 | H8 | 1.0990 | 2.2051 | 2.8246 | 4.1815 | 5.1757 | 6.2680 | 1.7735 | | 1.7772 | 3.1075 | 2.5532 | 2.6648 | 3.2072 | H9 | 1.0990 | 2.2051 | 2.8246 | 4.1815 | 5.1757 | 6.2680 | 1.7735 | 1.7772 | | 2.5532 | 3.1075 | 3.2072 | 2.6648 | H10 | 2.1884 | 1.0980 | 2.1607 | 2.6396 | 4.0130 | 4.7604 | 2.5252 | 3.1075 | 2.5532 | | 1.7655 | 3.0875 | 2.5235 | H11 | 2.1884 | 1.0980 | 2.1607 | 2.6396 | 4.0130 | 4.7604 | 2.5252 | 2.5532 | 3.1075 | 1.7655 | | 2.5235 | 3.0875 | H12 | 2.8362 | 2.1970 | 1.0976 | 2.1162 | 2.6593 | 3.7969 | 3.8372 | 2.6648 | 3.2072 | 3.0875 | 2.5235 | | 1.7924 | H13 | 2.8362 | 2.1970 | 1.0976 | 2.1162 | 2.6593 | 3.7969 | 3.8372 | 3.2072 | 2.6648 | 2.5235 | 3.0875 | 1.7924 | |
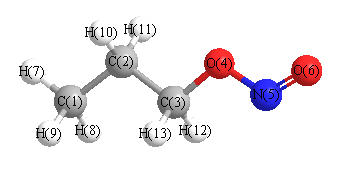 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
111.464 |
|
C1 |
C2 |
H10 |
110.191 |
| C1 |
C2 |
H11 |
110.191 |
|
C2 |
C1 |
H7 |
110.415 |
| C2 |
C1 |
H8 |
111.453 |
|
C2 |
C1 |
H9 |
111.453 |
| C2 |
C3 |
O4 |
106.722 |
|
C2 |
C3 |
H12 |
111.817 |
| C2 |
C3 |
H13 |
111.817 |
|
C3 |
C2 |
H10 |
108.933 |
| C3 |
C2 |
H11 |
108.933 |
|
C3 |
O4 |
N5 |
110.530 |
| O4 |
C3 |
H12 |
108.433 |
|
O4 |
C3 |
H13 |
108.433 |
| O4 |
N5 |
O6 |
110.693 |
|
H7 |
C1 |
H8 |
107.722 |
| H7 |
C1 |
H9 |
107.722 |
|
H8 |
C1 |
H9 |
107.912 |
| H10 |
C2 |
H11 |
107.011 |
|
H12 |
C3 |
H13 |
109.473 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-121G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.483 |
|
|
|
| 2 |
C |
-0.192 |
|
|
|
| 3 |
C |
-0.168 |
|
|
|
| 4 |
O |
-0.236 |
|
|
|
| 5 |
N |
0.115 |
|
|
|
| 6 |
O |
-0.101 |
|
|
|
| 7 |
H |
0.163 |
|
|
|
| 8 |
H |
0.147 |
|
|
|
| 9 |
H |
0.147 |
|
|
|
| 10 |
H |
0.154 |
|
|
|
| 11 |
H |
0.154 |
|
|
|
| 12 |
H |
0.150 |
|
|
|
| 13 |
H |
0.150 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-1.528 |
3.161 |
0.000 |
3.511 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.584 |
1.987 |
0.000 |
| y |
1.987 |
-41.299 |
0.000 |
| z |
0.000 |
0.000 |
-35.098 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.386 |
1.987 |
0.000 |
| y |
1.987 |
-4.458 |
0.000 |
| z |
0.000 |
0.000 |
4.844 |
|
| Polar |
|---|
| 3z2-r2 | 9.688 |
|---|
| x2-y2 | 2.715 |
|---|
| xy | 1.987 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.809 |
-2.604 |
0.000 |
| y |
-2.604 |
9.309 |
0.000 |
| z |
0.000 |
0.000 |
5.867 |
<r2> (average value of r
2) Å
2
| <r2> |
204.343 |
| (<r2>)1/2 |
14.295 |
Jump to
S1C1
Energy calculated at B3LYP/CEP-121G
| | hartrees |
|---|
| Energy at 0K | -62.839409 |
| Energy at 298.15K | -62.848244 |
| HF Energy | -62.839409 |
| Nuclear repulsion energy | 131.915232 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/CEP-121G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3091 |
3012 |
44.91 |
|
|
|
| 2 |
A |
3088 |
3009 |
46.95 |
|
|
|
| 3 |
A |
3075 |
2996 |
57.95 |
|
|
|
| 4 |
A |
3043 |
2966 |
14.28 |
|
|
|
| 5 |
A |
3017 |
2940 |
34.26 |
|
|
|
| 6 |
A |
2993 |
2916 |
16.30 |
|
|
|
| 7 |
A |
2988 |
2911 |
45.49 |
|
|
|
| 8 |
A |
1559 |
1520 |
200.71 |
|
|
|
| 9 |
A |
1530 |
1491 |
10.80 |
|
|
|
| 10 |
A |
1522 |
1483 |
8.84 |
|
|
|
| 11 |
A |
1503 |
1465 |
5.60 |
|
|
|
| 12 |
A |
1502 |
1464 |
4.48 |
|
|
|
| 13 |
A |
1434 |
1398 |
7.62 |
|
|
|
| 14 |
A |
1387 |
1352 |
5.25 |
|
|
|
| 15 |
A |
1385 |
1349 |
18.39 |
|
|
|
| 16 |
A |
1299 |
1266 |
0.91 |
|
|
|
| 17 |
A |
1270 |
1237 |
10.22 |
|
|
|
| 18 |
A |
1180 |
1150 |
7.15 |
|
|
|
| 19 |
A |
1113 |
1085 |
3.21 |
|
|
|
| 20 |
A |
1072 |
1045 |
11.17 |
|
|
|
| 21 |
A |
952 |
927 |
29.45 |
|
|
|
| 22 |
A |
932 |
908 |
50.37 |
|
|
|
| 23 |
A |
861 |
839 |
2.62 |
|
|
|
| 24 |
A |
815 |
794 |
350.47 |
|
|
|
| 25 |
A |
781 |
761 |
44.19 |
|
|
|
| 26 |
A |
574 |
559 |
7.00 |
|
|
|
| 27 |
A |
437 |
426 |
4.65 |
|
|
|
| 28 |
A |
347 |
339 |
1.98 |
|
|
|
| 29 |
A |
266 |
259 |
1.13 |
|
|
|
| 30 |
A |
225 |
220 |
1.08 |
|
|
|
| 31 |
A |
168 |
163 |
0.63 |
|
|
|
| 32 |
A |
100 |
98 |
1.68 |
|
|
|
| 33 |
A |
47 |
45 |
0.19 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22776.5 cm
-1
Scaled (by 0.9745) Zero Point Vibrational Energy (zpe) 22195.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/CEP-121G
Point Group is C1
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
-2.262 |
-0.943 |
0.183 |
| C2 |
-1.821 |
0.453 |
-0.324 |
| C3 |
-0.457 |
0.914 |
0.228 |
| O4 |
0.580 |
-0.018 |
-0.278 |
| N5 |
1.865 |
0.212 |
0.308 |
| O6 |
2.709 |
-0.561 |
-0.144 |
| H7 |
-3.244 |
-1.216 |
-0.224 |
| H8 |
-2.340 |
-0.962 |
1.279 |
| H9 |
-1.546 |
-1.716 |
-0.117 |
| H10 |
-1.779 |
0.462 |
-1.422 |
| H11 |
-2.564 |
1.211 |
-0.033 |
| H12 |
-0.208 |
1.927 |
-0.111 |
| H13 |
-0.442 |
0.896 |
1.326 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
O4 |
N5 |
O6 |
H7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
| C1 | | 1.5500 | 2.5902 | 3.0241 | 4.2878 | 4.9972 | 1.0971 | 1.0992 | 1.0956 | 2.1873 | 2.1860 | 3.5423 | 2.8295 |
C2 | 1.5500 | | 1.5418 | 2.4468 | 3.7475 | 4.6460 | 2.1958 | 2.2010 | 2.1965 | 1.0987 | 1.1003 | 2.1954 | 2.1959 | C3 | 2.5902 | 1.5418 | | 1.4826 | 2.4269 | 3.5127 | 3.5365 | 2.8582 | 2.8671 | 2.1620 | 2.1436 | 1.0977 | 1.0982 | O4 | 3.0241 | 2.4468 | 1.4826 | | 1.4311 | 2.2020 | 4.0074 | 3.4409 | 2.7255 | 2.6650 | 3.3839 | 2.1053 | 2.1099 | N5 | 4.2878 | 3.7475 | 2.4269 | 1.4311 | | 1.2310 | 5.3315 | 4.4725 | 3.9414 | 4.0415 | 4.5528 | 2.7231 | 2.6127 | O6 | 4.9972 | 4.6460 | 3.5127 | 2.2020 | 1.2310 | | 5.9900 | 5.2615 | 4.4096 | 4.7774 | 5.5641 | 3.8347 | 3.7705 | H7 | 1.0971 | 2.1958 | 3.5365 | 4.0074 | 5.3315 | 5.9900 | | 1.7725 | 1.7733 | 2.5293 | 2.5277 | 4.3716 | 3.8364 | H8 | 1.0992 | 2.2010 | 2.8582 | 3.4409 | 4.4725 | 5.2615 | 1.7725 | | 1.7744 | 3.1049 | 2.5487 | 3.8507 | 2.6571 | H9 | 1.0956 | 2.1965 | 2.8671 | 2.7255 | 3.9414 | 4.4096 | 1.7733 | 1.7744 | | 2.5496 | 3.1001 | 3.8814 | 3.1825 | H10 | 2.1873 | 1.0987 | 2.1620 | 2.6650 | 4.0415 | 4.7774 | 2.5293 | 3.1049 | 2.5496 | | 1.7625 | 2.5167 | 3.0869 | H11 | 2.1860 | 1.1003 | 2.1436 | 3.3839 | 4.5528 | 5.5641 | 2.5277 | 2.5487 | 3.1001 | 1.7625 | | 2.4634 | 2.5395 | H12 | 3.5423 | 2.1954 | 1.0977 | 2.1053 | 2.7231 | 3.8347 | 4.3716 | 3.8507 | 3.8814 | 2.5167 | 2.4634 | | 1.7840 | H13 | 2.8295 | 2.1959 | 1.0982 | 2.1099 | 2.6127 | 3.7705 | 3.8364 | 2.6571 | 3.1825 | 3.0869 | 2.5395 | 1.7840 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
113.810 |
|
C1 |
C2 |
H10 |
110.175 |
| C1 |
C2 |
H11 |
109.982 |
|
C2 |
C1 |
H7 |
110.939 |
| C2 |
C1 |
H8 |
111.222 |
|
C2 |
C1 |
H9 |
111.085 |
| C2 |
C3 |
O4 |
107.988 |
|
C2 |
C3 |
H12 |
111.450 |
| C2 |
C3 |
H13 |
111.450 |
|
C3 |
C2 |
H10 |
108.766 |
| C3 |
C2 |
H11 |
107.270 |
|
C3 |
O4 |
N5 |
112.792 |
| O4 |
C3 |
H12 |
108.443 |
|
O4 |
C3 |
H13 |
108.762 |
| O4 |
N5 |
O6 |
111.395 |
|
H7 |
C1 |
H8 |
107.606 |
| H7 |
C1 |
H9 |
107.944 |
|
H8 |
C1 |
H9 |
107.890 |
| H10 |
C2 |
H11 |
106.546 |
|
H12 |
C3 |
H13 |
108.667 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-121G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.486 |
|
|
|
| 2 |
C |
-0.187 |
|
|
|
| 3 |
C |
-0.165 |
|
|
|
| 4 |
O |
-0.233 |
|
|
|
| 5 |
N |
0.108 |
|
|
|
| 6 |
O |
-0.107 |
|
|
|
| 7 |
H |
0.155 |
|
|
|
| 8 |
H |
0.142 |
|
|
|
| 9 |
H |
0.162 |
|
|
|
| 10 |
H |
0.156 |
|
|
|
| 11 |
H |
0.139 |
|
|
|
| 12 |
H |
0.165 |
|
|
|
| 13 |
H |
0.150 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.993 |
-1.050 |
1.079 |
3.351 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-40.105 |
0.615 |
-0.978 |
| y |
0.615 |
-35.028 |
-0.976 |
| z |
-0.978 |
-0.976 |
-37.857 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-3.663 |
0.615 |
-0.978 |
| y |
0.615 |
3.953 |
-0.976 |
| z |
-0.978 |
-0.976 |
-0.291 |
|
| Polar |
|---|
| 3z2-r2 | -0.581 |
|---|
| x2-y2 | -5.077 |
|---|
| xy | 0.615 |
|---|
| xz | -0.978 |
|---|
| yz | -0.976 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.256 |
-1.420 |
0.007 |
| y |
-1.420 |
7.093 |
-0.023 |
| z |
0.007 |
-0.023 |
6.306 |
<r2> (average value of r
2) Å
2
| <r2> |
166.804 |
| (<r2>)1/2 |
12.915 |