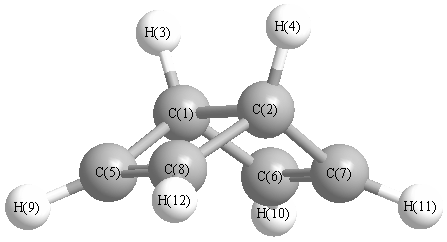
Vibrational Frequencies calculated at B3LYP/CEP-121G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3181 |
3084 |
14.36 |
324.61 |
0.04 |
0.07 |
| 2 |
A1 |
3082 |
2988 |
54.76 |
168.48 |
0.14 |
0.24 |
| 3 |
A1 |
1596 |
1547 |
0.10 |
37.29 |
0.05 |
0.09 |
| 4 |
A1 |
1170 |
1135 |
5.10 |
27.74 |
0.14 |
0.25 |
| 5 |
A1 |
1028 |
997 |
1.91 |
13.68 |
0.70 |
0.82 |
| 6 |
A1 |
934 |
906 |
10.65 |
10.68 |
0.26 |
0.41 |
| 7 |
A1 |
859 |
832 |
0.08 |
6.32 |
0.72 |
0.83 |
| 8 |
A1 |
799 |
775 |
86.04 |
7.22 |
0.06 |
0.12 |
| 9 |
A1 |
383 |
371 |
5.46 |
10.96 |
0.60 |
0.75 |
| 10 |
A2 |
3148 |
3052 |
0.00 |
202.86 |
0.75 |
0.86 |
| 11 |
A2 |
1278 |
1239 |
0.00 |
8.19 |
0.75 |
0.86 |
| 12 |
A2 |
1193 |
1156 |
0.00 |
11.30 |
0.75 |
0.86 |
| 13 |
A2 |
938 |
909 |
0.00 |
0.54 |
0.75 |
0.86 |
| 14 |
A2 |
912 |
884 |
0.00 |
1.48 |
0.75 |
0.86 |
| 15 |
A2 |
783 |
759 |
0.00 |
2.60 |
0.75 |
0.86 |
| 16 |
A2 |
332 |
322 |
0.00 |
2.45 |
0.75 |
0.86 |
| 17 |
B1 |
3178 |
3081 |
64.34 |
78.05 |
0.75 |
0.86 |
| 18 |
B1 |
1568 |
1520 |
15.19 |
1.55 |
0.75 |
0.86 |
| 19 |
B1 |
1203 |
1167 |
4.30 |
1.93 |
0.75 |
0.86 |
| 20 |
B1 |
1087 |
1053 |
0.22 |
0.00 |
0.75 |
0.86 |
| 21 |
B1 |
983 |
953 |
0.82 |
8.90 |
0.75 |
0.86 |
| 22 |
B1 |
709 |
687 |
51.25 |
4.07 |
0.75 |
0.86 |
| 23 |
B2 |
3150 |
3053 |
49.15 |
53.12 |
0.75 |
0.86 |
| 24 |
B2 |
3074 |
2980 |
26.13 |
143.10 |
0.75 |
0.86 |
| 25 |
B2 |
1285 |
1246 |
37.91 |
0.01 |
0.75 |
0.86 |
| 26 |
B2 |
1149 |
1113 |
4.28 |
0.73 |
0.75 |
0.86 |
| 27 |
B2 |
943 |
914 |
7.08 |
0.34 |
0.75 |
0.86 |
| 28 |
B2 |
926 |
898 |
2.53 |
0.93 |
0.75 |
0.86 |
| 29 |
B2 |
820 |
795 |
10.22 |
1.26 |
0.75 |
0.86 |
| 30 |
B2 |
483 |
468 |
5.59 |
5.26 |
0.75 |
0.86 |
Unscaled Zero Point Vibrational Energy (zpe) 21086.1 cm
-1
Scaled (by 0.9694) Zero Point Vibrational Energy (zpe) 20440.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-121G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.134 |
|
|
|
| 2 |
C |
-0.134 |
|
|
|
| 3 |
H |
0.186 |
|
|
|
| 4 |
H |
0.186 |
|
|
|
| 5 |
C |
-0.221 |
|
|
|
| 6 |
C |
-0.221 |
|
|
|
| 7 |
C |
-0.221 |
|
|
|
| 8 |
C |
-0.221 |
|
|
|
| 9 |
H |
0.195 |
|
|
|
| 10 |
H |
0.195 |
|
|
|
| 11 |
H |
0.195 |
|
|
|
| 12 |
H |
0.195 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.050 |
0.050 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.722 |
0.000 |
0.000 |
| y |
0.000 |
-32.703 |
0.000 |
| z |
0.000 |
0.000 |
-36.217 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.261 |
0.000 |
0.000 |
| y |
0.000 |
3.266 |
0.000 |
| z |
0.000 |
0.000 |
-2.005 |
|
| Polar |
|---|
| 3z2-r2 | -4.009 |
|---|
| x2-y2 | -3.018 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.701 |
0.000 |
0.000 |
| y |
0.000 |
10.269 |
0.000 |
| z |
0.000 |
0.000 |
6.879 |
<r2> (average value of r
2) Å
2
| <r2> |
98.901 |
| (<r2>)1/2 |
9.945 |