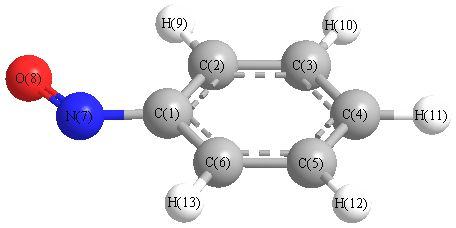
Vibrational Frequencies calculated at B3LYP/CEP-121G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3199 |
3101 |
5.75 |
|
|
|
| 2 |
A' |
3188 |
3090 |
16.34 |
|
|
|
| 3 |
A' |
3178 |
3081 |
20.99 |
|
|
|
| 4 |
A' |
3170 |
3073 |
13.93 |
|
|
|
| 5 |
A' |
3158 |
3062 |
0.92 |
|
|
|
| 6 |
A' |
1624 |
1574 |
6.58 |
|
|
|
| 7 |
A' |
1614 |
1564 |
2.98 |
|
|
|
| 8 |
A' |
1557 |
1509 |
187.95 |
|
|
|
| 9 |
A' |
1485 |
1439 |
13.63 |
|
|
|
| 10 |
A' |
1465 |
1420 |
39.92 |
|
|
|
| 11 |
A' |
1339 |
1298 |
14.85 |
|
|
|
| 12 |
A' |
1321 |
1280 |
8.37 |
|
|
|
| 13 |
A' |
1185 |
1149 |
15.43 |
|
|
|
| 14 |
A' |
1167 |
1131 |
1.38 |
|
|
|
| 15 |
A' |
1120 |
1085 |
145.60 |
|
|
|
| 16 |
A' |
1081 |
1048 |
4.84 |
|
|
|
| 17 |
A' |
1020 |
989 |
6.13 |
|
|
|
| 18 |
A' |
999 |
968 |
1.99 |
|
|
|
| 19 |
A' |
820 |
795 |
30.50 |
|
|
|
| 20 |
A' |
667 |
646 |
12.35 |
|
|
|
| 21 |
A' |
613 |
594 |
0.15 |
|
|
|
| 22 |
A' |
440 |
426 |
0.76 |
|
|
|
| 23 |
A' |
255 |
247 |
2.14 |
|
|
|
| 24 |
A" |
1026 |
994 |
0.60 |
|
|
|
| 25 |
A" |
1005 |
974 |
0.03 |
|
|
|
| 26 |
A" |
973 |
943 |
4.28 |
|
|
|
| 27 |
A" |
874 |
847 |
0.00 |
|
|
|
| 28 |
A" |
787 |
763 |
64.68 |
|
|
|
| 29 |
A" |
690 |
669 |
24.95 |
|
|
|
| 30 |
A" |
476 |
462 |
2.32 |
|
|
|
| 31 |
A" |
419 |
406 |
0.00 |
|
|
|
| 32 |
A" |
251 |
243 |
0.14 |
|
|
|
| 33 |
A" |
116 |
113 |
0.17 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 21137.6 cm
-1
Scaled (by 0.9694) Zero Point Vibrational Energy (zpe) 20490.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/CEP-121G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.001 |
|
|
|
| 2 |
C |
-0.299 |
|
|
|
| 3 |
C |
-0.157 |
|
|
|
| 4 |
C |
-0.207 |
|
|
|
| 5 |
C |
-0.178 |
|
|
|
| 6 |
C |
-0.241 |
|
|
|
| 7 |
N |
0.262 |
|
|
|
| 8 |
O |
-0.271 |
|
|
|
| 9 |
H |
0.226 |
|
|
|
| 10 |
H |
0.219 |
|
|
|
| 11 |
H |
0.213 |
|
|
|
| 12 |
H |
0.221 |
|
|
|
| 13 |
H |
0.213 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.230 |
-3.744 |
0.000 |
3.941 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.500 |
2.593 |
0.000 |
| y |
2.593 |
-48.566 |
0.000 |
| z |
0.000 |
0.000 |
-47.875 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.721 |
2.593 |
0.000 |
| y |
2.593 |
-3.879 |
0.000 |
| z |
0.000 |
0.000 |
-2.842 |
|
| Polar |
|---|
| 3z2-r2 | -5.684 |
|---|
| x2-y2 | 7.066 |
|---|
| xy | 2.593 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
13.256 |
-1.837 |
0.000 |
| y |
-1.837 |
16.025 |
0.000 |
| z |
0.000 |
0.000 |
5.278 |
<r2> (average value of r
2) Å
2
| <r2> |
197.552 |
| (<r2>)1/2 |
14.055 |