Jump to
S1C2
Energy calculated at B3LYP/LANL2DZ
| | hartrees |
|---|
| Energy at 0K | -68.507704 |
| Energy at 298.15K | |
| HF Energy | -68.507704 |
| Nuclear repulsion energy | 39.389667 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/LANL2DZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3313 |
3184 |
0.58 |
|
|
|
| 2 |
A1 |
678 |
652 |
17.09 |
|
|
|
| 3 |
A1 |
286 |
275 |
0.50 |
|
|
|
| 4 |
B1 |
411i |
395i |
118.81 |
|
|
|
| 5 |
B2 |
1213 |
1166 |
40.48 |
|
|
|
| 6 |
B2 |
845 |
812 |
171.34 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 2961.7 cm
-1
Scaled (by 0.9612) Zero Point Vibrational Energy (zpe) 2846.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/LANL2DZ
Point Group is C2v
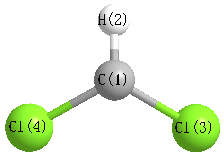
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.704 |
| H2 |
0.000 |
0.000 |
1.784 |
| Cl3 |
0.000 |
1.540 |
-0.177 |
| Cl4 |
0.000 |
-1.540 |
-0.177 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
Cl3 |
Cl4 |
| C1 | | 1.0799 | 1.7740 | 1.7740 |
H2 | 1.0799 | | 2.4933 | 2.4933 | Cl3 | 1.7740 | 2.4933 | | 3.0792 | Cl4 | 1.7740 | 2.4933 | 3.0792 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Cl3 |
C1 |
H2 |
119.788 |
|
Cl3 |
C1 |
Cl4 |
120.424 |
| Cl4 |
C1 |
H2 |
119.788 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/LANL2DZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.354 |
|
|
|
| 2 |
H |
0.290 |
|
|
|
| 3 |
Cl |
0.032 |
|
|
|
| 4 |
Cl |
0.032 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.248 |
1.248 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.671 |
0.000 |
0.000 |
| y |
0.000 |
-30.770 |
0.000 |
| z |
0.000 |
0.000 |
-27.271 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.650 |
0.000 |
0.000 |
| y |
0.000 |
-1.799 |
0.000 |
| z |
0.000 |
0.000 |
3.450 |
|
| Polar |
|---|
| 3z2-r2 | 6.900 |
|---|
| x2-y2 | 0.099 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.126 |
0.000 |
0.000 |
| y |
0.000 |
6.237 |
0.000 |
| z |
0.000 |
0.000 |
2.714 |
<r2> (average value of r
2) Å
2
| <r2> |
58.253 |
| (<r2>)1/2 |
7.632 |
Jump to
S1C1
Energy calculated at B3LYP/LANL2DZ
| | hartrees |
|---|
| Energy at 0K | -68.509116 |
| Energy at 298.15K | -68.509840 |
| HF Energy | -68.509116 |
| Nuclear repulsion energy | 39.209304 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/LANL2DZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3257 |
3130 |
3.84 |
|
|
|
| 2 |
A' |
701 |
674 |
32.56 |
|
|
|
| 3 |
A' |
515 |
495 |
27.12 |
|
|
|
| 4 |
A' |
281 |
270 |
0.55 |
|
|
|
| 5 |
A" |
1224 |
1176 |
31.65 |
|
|
|
| 6 |
A" |
801 |
770 |
196.29 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 3389.2 cm
-1
Scaled (by 0.9612) Zero Point Vibrational Energy (zpe) 3257.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/LANL2DZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.014 |
0.738 |
0.000 |
| H2 |
-0.559 |
1.660 |
0.000 |
| Cl3 |
0.014 |
-0.179 |
1.537 |
| Cl4 |
0.014 |
-0.179 |
-1.537 |
Atom - Atom Distances (Å)
| |
C1 |
H2 |
Cl3 |
Cl4 |
| C1 | | 1.0849 | 1.7896 | 1.7896 |
H2 | 1.0849 | | 2.4638 | 2.4638 | Cl3 | 1.7896 | 2.4638 | | 3.0732 | Cl4 | 1.7896 | 2.4638 | 3.0732 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| Cl3 |
C1 |
H2 |
115.810 |
|
Cl3 |
C1 |
Cl4 |
118.323 |
| Cl4 |
C1 |
H2 |
115.810 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/LANL2DZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.316 |
|
|
|
| 2 |
H |
0.279 |
|
|
|
| 3 |
Cl |
0.018 |
|
|
|
| 4 |
Cl |
0.018 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.809 |
1.131 |
0.000 |
1.390 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.237 |
-1.218 |
0.000 |
| y |
-1.218 |
-27.903 |
0.000 |
| z |
0.000 |
0.000 |
-31.113 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.729 |
-1.218 |
0.000 |
| y |
-1.218 |
2.772 |
0.000 |
| z |
0.000 |
0.000 |
-2.043 |
|
| Polar |
|---|
| 3z2-r2 | -4.086 |
|---|
| x2-y2 | -2.334 |
|---|
| xy | -1.218 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
1.211 |
-0.173 |
0.000 |
| y |
-0.173 |
2.815 |
0.000 |
| z |
0.000 |
0.000 |
6.341 |
<r2> (average value of r
2) Å
2
| <r2> |
58.427 |
| (<r2>)1/2 |
7.644 |