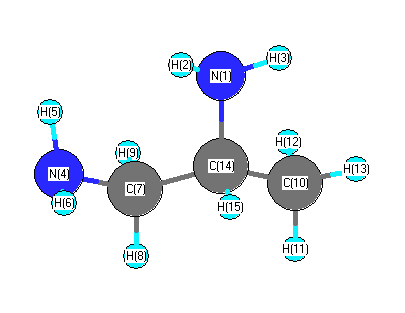
Vibrational Frequencies calculated at B3LYP/LANL2DZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3720 |
3575 |
3.77 |
|
|
|
| 2 |
A |
3655 |
3514 |
0.97 |
|
|
|
| 3 |
A |
3584 |
3445 |
0.96 |
|
|
|
| 4 |
A |
3534 |
3396 |
1.75 |
|
|
|
| 5 |
A |
3137 |
3015 |
60.12 |
|
|
|
| 6 |
A |
3117 |
2996 |
52.11 |
|
|
|
| 7 |
A |
3113 |
2993 |
28.50 |
|
|
|
| 8 |
A |
3045 |
2927 |
63.65 |
|
|
|
| 9 |
A |
3033 |
2915 |
25.70 |
|
|
|
| 10 |
A |
2914 |
2801 |
114.76 |
|
|
|
| 11 |
A |
1676 |
1611 |
63.43 |
|
|
|
| 12 |
A |
1672 |
1607 |
32.46 |
|
|
|
| 13 |
A |
1523 |
1464 |
7.08 |
|
|
|
| 14 |
A |
1513 |
1454 |
9.51 |
|
|
|
| 15 |
A |
1493 |
1435 |
3.31 |
|
|
|
| 16 |
A |
1430 |
1374 |
19.95 |
|
|
|
| 17 |
A |
1412 |
1357 |
0.87 |
|
|
|
| 18 |
A |
1395 |
1341 |
21.37 |
|
|
|
| 19 |
A |
1364 |
1311 |
2.54 |
|
|
|
| 20 |
A |
1350 |
1298 |
4.25 |
|
|
|
| 21 |
A |
1256 |
1207 |
1.91 |
|
|
|
| 22 |
A |
1196 |
1149 |
3.77 |
|
|
|
| 23 |
A |
1185 |
1139 |
8.09 |
|
|
|
| 24 |
A |
1120 |
1077 |
39.82 |
|
|
|
| 25 |
A |
1037 |
997 |
8.51 |
|
|
|
| 26 |
A |
1016 |
976 |
1.19 |
|
|
|
| 27 |
A |
954 |
917 |
2.39 |
|
|
|
| 28 |
A |
877 |
843 |
4.32 |
|
|
|
| 29 |
A |
836 |
804 |
22.13 |
|
|
|
| 30 |
A |
650 |
625 |
196.18 |
|
|
|
| 31 |
A |
509 |
489 |
57.13 |
|
|
|
| 32 |
A |
470 |
451 |
38.22 |
|
|
|
| 33 |
A |
381 |
366 |
181.74 |
|
|
|
| 34 |
A |
365 |
351 |
3.94 |
|
|
|
| 35 |
A |
317 |
304 |
137.42 |
|
|
|
| 36 |
A |
254 |
244 |
14.75 |
|
|
|
| 37 |
A |
217 |
209 |
5.51 |
|
|
|
| 38 |
A |
195 |
188 |
120.51 |
|
|
|
| 39 |
A |
121 |
117 |
2.20 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30317.9 cm
-1
Scaled (by 0.9612) Zero Point Vibrational Energy (zpe) 29141.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/LANL2DZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.609 |
|
|
|
| 2 |
H |
0.249 |
|
|
|
| 3 |
H |
0.261 |
|
|
|
| 4 |
N |
-0.605 |
|
|
|
| 5 |
H |
0.274 |
|
|
|
| 6 |
H |
0.262 |
|
|
|
| 7 |
C |
-0.322 |
|
|
|
| 8 |
H |
0.177 |
|
|
|
| 9 |
H |
0.201 |
|
|
|
| 10 |
C |
-0.653 |
|
|
|
| 11 |
H |
0.191 |
|
|
|
| 12 |
H |
0.213 |
|
|
|
| 13 |
H |
0.190 |
|
|
|
| 14 |
C |
0.019 |
|
|
|
| 15 |
H |
0.154 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.488 |
1.075 |
1.601 |
1.989 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.571 |
-0.241 |
-0.076 |
| y |
-0.241 |
6.795 |
-0.263 |
| z |
-0.076 |
-0.263 |
5.919 |
<r2> (average value of r
2) Å
2
| <r2> |
146.535 |
| (<r2>)1/2 |
12.105 |