Jump to
S1C2
Vibrational Frequencies calculated at B3LYP/SDD
Geometric Data calculated at B3LYP/SDD
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Jump to
S1C1
Energy calculated at B3LYP/SDD
| | hartrees |
|---|
| Energy at 0K | -485.249720 |
| Energy at 298.15K | -485.253392 |
| Nuclear repulsion energy | 304.658081 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/SDD
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
1616 |
1553 |
259.53 |
|
|
|
| 2 |
A |
1251 |
1203 |
45.34 |
|
|
|
| 3 |
A |
814 |
783 |
23.39 |
|
|
|
| 4 |
A |
718 |
691 |
1.50 |
|
|
|
| 5 |
A |
595 |
572 |
0.23 |
|
|
|
| 6 |
A |
362 |
348 |
0.37 |
|
|
|
| 7 |
A |
187 |
180 |
0.59 |
|
|
|
| 8 |
A |
61 |
59 |
0.25 |
|
|
|
| 9 |
B |
1578 |
1517 |
265.21 |
|
|
|
| 10 |
B |
1163 |
1118 |
334.65 |
|
|
|
| 11 |
B |
687 |
660 |
408.79 |
|
|
|
| 12 |
B |
643 |
618 |
22.32 |
|
|
|
| 13 |
B |
555 |
533 |
300.49 |
|
|
|
| 14 |
B |
364 |
350 |
154.41 |
|
|
|
| 15 |
B |
42 |
41 |
0.27 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 5318.2 cm
-1
Scaled (by 0.9613) Zero Point Vibrational Energy (zpe) 5112.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/SDD
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.869 |
| N2 |
0.000 |
1.311 |
0.015 |
| N3 |
0.000 |
-1.311 |
0.015 |
| O4 |
0.594 |
2.219 |
0.612 |
| O5 |
-0.594 |
-2.219 |
0.612 |
| O6 |
-0.622 |
1.268 |
-1.060 |
| O7 |
0.622 |
-1.268 |
-1.060 |
Atom - Atom Distances (Å)
| |
O1 |
N2 |
N3 |
O4 |
O5 |
O6 |
O7 |
| O1 | | 1.5648 | 1.5648 | 2.3111 | 2.3111 | 2.3907 | 2.3907 |
N2 | 1.5648 | | 2.6217 | 1.2385 | 3.6286 | 1.2422 | 2.8623 | N3 | 1.5648 | 2.6217 | | 3.6286 | 1.2385 | 2.8623 | 1.2422 | O4 | 2.3111 | 1.2385 | 3.6286 | | 4.5935 | 2.2755 | 3.8667 | O5 | 2.3111 | 3.6286 | 1.2385 | 4.5935 | | 3.8667 | 2.2755 | O6 | 2.3907 | 1.2422 | 2.8623 | 2.2755 | 3.8667 | | 2.8251 | O7 | 2.3907 | 2.8623 | 1.2422 | 3.8667 | 2.2755 | 2.8251 | |
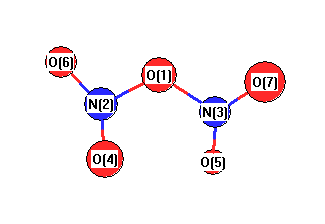 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
N2 |
O4 |
110.521 |
|
O1 |
N2 |
O6 |
116.324 |
| O1 |
N3 |
O5 |
110.521 |
|
O1 |
N3 |
O7 |
116.324 |
| N2 |
O1 |
N3 |
113.801 |
|
O4 |
N2 |
O6 |
133.056 |
| O5 |
N3 |
O7 |
133.056 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/SDD
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.153 |
|
|
|
| 2 |
N |
0.274 |
|
|
|
| 3 |
N |
0.274 |
|
|
|
| 4 |
O |
-0.094 |
|
|
|
| 5 |
O |
-0.094 |
|
|
|
| 6 |
O |
-0.104 |
|
|
|
| 7 |
O |
-0.104 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.022 |
0.022 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-37.477 |
-1.583 |
0.000 |
| y |
-1.583 |
-40.753 |
0.000 |
| z |
0.000 |
0.000 |
-41.315 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.557 |
-1.583 |
0.000 |
| y |
-1.583 |
-1.357 |
0.000 |
| z |
0.000 |
0.000 |
-2.200 |
|
| Polar |
|---|
| 3z2-r2 | -4.399 |
|---|
| x2-y2 | 3.276 |
|---|
| xy | -1.583 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
3.496 |
1.219 |
0.000 |
| y |
1.219 |
8.942 |
0.000 |
| z |
0.000 |
0.000 |
5.668 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |