Vibrational Frequencies calculated at B3LYP/SDD
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3169 |
3047 |
33.79 |
|
|
|
| 2 |
A |
3145 |
3023 |
52.78 |
|
|
|
| 3 |
A |
3121 |
3000 |
59.88 |
|
|
|
| 4 |
A |
3117 |
2997 |
39.49 |
|
|
|
| 5 |
A |
3106 |
2986 |
21.48 |
|
|
|
| 6 |
A |
3093 |
2973 |
35.47 |
|
|
|
| 7 |
A |
3083 |
2964 |
5.63 |
|
|
|
| 8 |
A |
3055 |
2937 |
35.64 |
|
|
|
| 9 |
A |
3044 |
2926 |
47.17 |
|
|
|
| 10 |
A |
3034 |
2916 |
29.16 |
|
|
|
| 11 |
A |
1521 |
1462 |
10.34 |
|
|
|
| 12 |
A |
1517 |
1458 |
2.56 |
|
|
|
| 13 |
A |
1511 |
1452 |
13.27 |
|
|
|
| 14 |
A |
1508 |
1449 |
9.49 |
|
|
|
| 15 |
A |
1504 |
1446 |
3.72 |
|
|
|
| 16 |
A |
1438 |
1382 |
11.25 |
|
|
|
| 17 |
A |
1381 |
1328 |
2.86 |
|
|
|
| 18 |
A |
1369 |
1316 |
1.83 |
|
|
|
| 19 |
A |
1339 |
1287 |
3.62 |
|
|
|
| 20 |
A |
1314 |
1263 |
6.30 |
|
|
|
| 21 |
A |
1292 |
1242 |
25.04 |
|
|
|
| 22 |
A |
1243 |
1195 |
5.39 |
|
|
|
| 23 |
A |
1210 |
1163 |
7.55 |
|
|
|
| 24 |
A |
1165 |
1120 |
0.22 |
|
|
|
| 25 |
A |
1122 |
1079 |
0.07 |
|
|
|
| 26 |
A |
1085 |
1043 |
2.04 |
|
|
|
| 27 |
A |
1062 |
1021 |
9.30 |
|
|
|
| 28 |
A |
1041 |
1001 |
4.90 |
|
|
|
| 29 |
A |
990 |
951 |
7.03 |
|
|
|
| 30 |
A |
964 |
926 |
5.79 |
|
|
|
| 31 |
A |
901 |
866 |
2.44 |
|
|
|
| 32 |
A |
877 |
843 |
4.32 |
|
|
|
| 33 |
A |
850 |
817 |
3.30 |
|
|
|
| 34 |
A |
646 |
621 |
2.98 |
|
|
|
| 35 |
A |
587 |
564 |
4.46 |
|
|
|
| 36 |
A |
522 |
502 |
1.36 |
|
|
|
| 37 |
A |
453 |
435 |
0.13 |
|
|
|
| 38 |
A |
405 |
389 |
1.15 |
|
|
|
| 39 |
A |
283 |
272 |
1.87 |
|
|
|
| 40 |
A |
247 |
237 |
0.28 |
|
|
|
| 41 |
A |
226 |
217 |
0.10 |
|
|
|
| 42 |
A |
92 |
89 |
1.25 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 31313.9 cm
-1
Scaled (by 0.9613) Zero Point Vibrational Energy (zpe) 30102.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
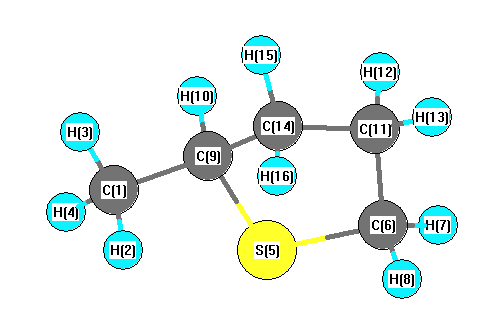
Charges from optimized geometry at B3LYP/SDD
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.615 |
|
|
|
| 2 |
H |
0.212 |
|
|
|
| 3 |
H |
0.191 |
|
|
|
| 4 |
H |
0.211 |
|
|
|
| 5 |
S |
0.129 |
|
|
|
| 6 |
C |
-0.629 |
|
|
|
| 7 |
H |
0.236 |
|
|
|
| 8 |
H |
0.235 |
|
|
|
| 9 |
C |
-0.341 |
|
|
|
| 10 |
H |
0.236 |
|
|
|
| 11 |
C |
-0.331 |
|
|
|
| 12 |
H |
0.194 |
|
|
|
| 13 |
H |
0.206 |
|
|
|
| 14 |
C |
-0.324 |
|
|
|
| 15 |
H |
0.204 |
|
|
|
| 16 |
H |
0.185 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.340 |
2.375 |
0.029 |
2.400 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-42.007 |
-0.205 |
1.040 |
| y |
-0.205 |
-48.406 |
-0.415 |
| z |
1.040 |
-0.415 |
-45.538 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.965 |
-0.205 |
1.040 |
| y |
-0.205 |
-4.634 |
-0.415 |
| z |
1.040 |
-0.415 |
-0.331 |
|
| Polar |
|---|
| 3z2-r2 | -0.663 |
|---|
| x2-y2 | 6.399 |
|---|
| xy | -0.205 |
|---|
| xz | 1.040 |
|---|
| yz | -0.415 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.665 |
-0.194 |
0.485 |
| y |
-0.194 |
10.116 |
-0.099 |
| z |
0.485 |
-0.099 |
7.538 |
<r2> (average value of r
2) Å
2
| <r2> |
210.055 |
| (<r2>)1/2 |
14.493 |