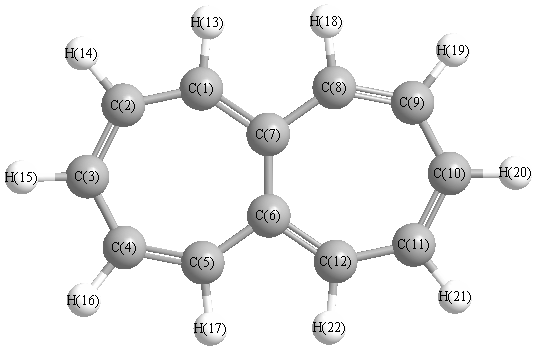
Vibrational Frequencies calculated at B3LYP/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3169 |
3066 |
2.41 |
|
|
|
| 2 |
A |
3150 |
3047 |
15.39 |
|
|
|
| 3 |
A |
3145 |
3043 |
0.41 |
|
|
|
| 4 |
A |
3129 |
3027 |
0.02 |
|
|
|
| 5 |
A |
3127 |
3025 |
0.35 |
|
|
|
| 6 |
A |
1691 |
1636 |
1.91 |
|
|
|
| 7 |
A |
1630 |
1577 |
3.74 |
|
|
|
| 8 |
A |
1576 |
1525 |
1.76 |
|
|
|
| 9 |
A |
1479 |
1431 |
0.02 |
|
|
|
| 10 |
A |
1453 |
1406 |
0.02 |
|
|
|
| 11 |
A |
1416 |
1370 |
1.03 |
|
|
|
| 12 |
A |
1272 |
1231 |
0.02 |
|
|
|
| 13 |
A |
1263 |
1222 |
0.03 |
|
|
|
| 14 |
A |
1219 |
1180 |
0.35 |
|
|
|
| 15 |
A |
1061 |
1027 |
1.09 |
|
|
|
| 16 |
A |
1014 |
981 |
0.41 |
|
|
|
| 17 |
A |
1005 |
973 |
0.16 |
|
|
|
| 18 |
A |
966 |
935 |
0.35 |
|
|
|
| 19 |
A |
911 |
882 |
13.40 |
|
|
|
| 20 |
A |
899 |
870 |
0.87 |
|
|
|
| 21 |
A |
829 |
802 |
4.11 |
|
|
|
| 22 |
A |
788 |
762 |
0.46 |
|
|
|
| 23 |
A |
731 |
707 |
73.43 |
|
|
|
| 24 |
A |
653 |
632 |
2.67 |
|
|
|
| 25 |
A |
585 |
566 |
0.08 |
|
|
|
| 26 |
A |
431 |
417 |
6.43 |
|
|
|
| 27 |
A |
389 |
376 |
5.18 |
|
|
|
| 28 |
A |
362 |
350 |
1.09 |
|
|
|
| 29 |
A |
299 |
289 |
2.18 |
|
|
|
| 30 |
A |
128 |
123 |
1.16 |
|
|
|
| 31 |
A |
71 |
69 |
0.01 |
|
|
|
| 32 |
B |
3168 |
3065 |
56.36 |
|
|
|
| 33 |
B |
3150 |
3048 |
48.86 |
|
|
|
| 34 |
B |
3144 |
3042 |
3.39 |
|
|
|
| 35 |
B |
3130 |
3029 |
2.08 |
|
|
|
| 36 |
B |
3126 |
3024 |
17.65 |
|
|
|
| 37 |
B |
1662 |
1608 |
10.20 |
|
|
|
| 38 |
B |
1631 |
1578 |
91.00 |
|
|
|
| 39 |
B |
1556 |
1506 |
27.95 |
|
|
|
| 40 |
B |
1483 |
1435 |
3.87 |
|
|
|
| 41 |
B |
1423 |
1377 |
4.93 |
|
|
|
| 42 |
B |
1348 |
1304 |
2.26 |
|
|
|
| 43 |
B |
1276 |
1234 |
12.04 |
|
|
|
| 44 |
B |
1251 |
1211 |
0.75 |
|
|
|
| 45 |
B |
1126 |
1089 |
2.49 |
|
|
|
| 46 |
B |
1048 |
1014 |
2.44 |
|
|
|
| 47 |
B |
1011 |
978 |
0.81 |
|
|
|
| 48 |
B |
1004 |
972 |
2.95 |
|
|
|
| 49 |
B |
943 |
912 |
5.40 |
|
|
|
| 50 |
B |
927 |
897 |
1.24 |
|
|
|
| 51 |
B |
886 |
857 |
13.16 |
|
|
|
| 52 |
B |
820 |
793 |
51.30 |
|
|
|
| 53 |
B |
753 |
729 |
12.36 |
|
|
|
| 54 |
B |
666 |
644 |
9.54 |
|
|
|
| 55 |
B |
598 |
579 |
14.57 |
|
|
|
| 56 |
B |
589 |
570 |
9.88 |
|
|
|
| 57 |
B |
428 |
414 |
1.14 |
|
|
|
| 58 |
B |
361 |
349 |
1.23 |
|
|
|
| 59 |
B |
286 |
277 |
0.57 |
|
|
|
| 60 |
B |
161 |
156 |
2.96 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 39396.7 cm
-1
Scaled (by 0.9675) Zero Point Vibrational Energy (zpe) 38116.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.