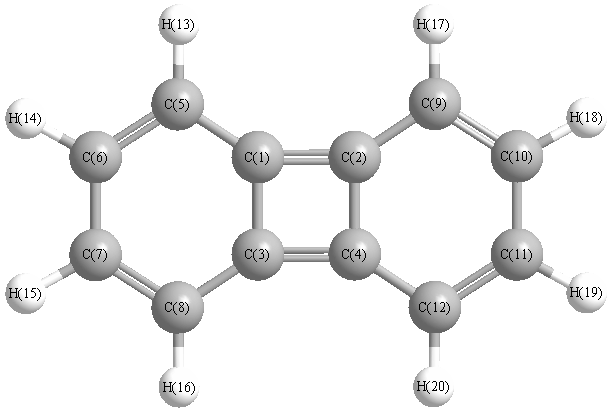
Vibrational Frequencies calculated at B3LYP/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
3193 |
3089 |
0.00 |
|
|
|
| 2 |
Ag |
3177 |
3074 |
0.00 |
|
|
|
| 3 |
Ag |
1701 |
1646 |
0.00 |
|
|
|
| 4 |
Ag |
1499 |
1451 |
0.00 |
|
|
|
| 5 |
Ag |
1436 |
1389 |
0.00 |
|
|
|
| 6 |
Ag |
1190 |
1152 |
0.00 |
|
|
|
| 7 |
Ag |
1132 |
1095 |
0.00 |
|
|
|
| 8 |
Ag |
1013 |
980 |
0.00 |
|
|
|
| 9 |
Ag |
783 |
757 |
0.00 |
|
|
|
| 10 |
Ag |
400 |
387 |
0.00 |
|
|
|
| 11 |
Au |
998 |
966 |
0.00 |
|
|
|
| 12 |
Au |
909 |
880 |
0.00 |
|
|
|
| 13 |
Au |
827 |
800 |
0.00 |
|
|
|
| 14 |
Au |
595 |
575 |
0.00 |
|
|
|
| 15 |
Au |
149 |
144 |
0.00 |
|
|
|
| 16 |
B1g |
999 |
966 |
0.00 |
|
|
|
| 17 |
B1g |
893 |
864 |
0.00 |
|
|
|
| 18 |
B1g |
732 |
708 |
0.00 |
|
|
|
| 19 |
B1g |
468 |
453 |
0.00 |
|
|
|
| 20 |
B1u |
3192 |
3088 |
35.26 |
|
|
|
| 21 |
B1u |
3177 |
3073 |
10.22 |
|
|
|
| 22 |
B1u |
1629 |
1576 |
0.01 |
|
|
|
| 23 |
B1u |
1463 |
1415 |
30.25 |
|
|
|
| 24 |
B1u |
1298 |
1256 |
24.34 |
|
|
|
| 25 |
B1u |
1179 |
1141 |
12.45 |
|
|
|
| 26 |
B1u |
1048 |
1014 |
0.01 |
|
|
|
| 27 |
B1u |
990 |
958 |
27.29 |
|
|
|
| 28 |
B1u |
629 |
609 |
3.68 |
|
|
|
| 29 |
B2g |
950 |
919 |
0.00 |
|
|
|
| 30 |
B2g |
765 |
740 |
0.00 |
|
|
|
| 31 |
B2g |
437 |
423 |
0.00 |
|
|
|
| 32 |
B2g |
319 |
309 |
0.00 |
|
|
|
| 33 |
B2u |
3186 |
3083 |
43.06 |
|
|
|
| 34 |
B2u |
3166 |
3063 |
0.11 |
|
|
|
| 35 |
B2u |
1621 |
1569 |
1.37 |
|
|
|
| 36 |
B2u |
1480 |
1432 |
4.95 |
|
|
|
| 37 |
B2u |
1296 |
1253 |
3.11 |
|
|
|
| 38 |
B2u |
1149 |
1111 |
13.65 |
|
|
|
| 39 |
B2u |
1077 |
1042 |
0.19 |
|
|
|
| 40 |
B2u |
736 |
713 |
0.25 |
|
|
|
| 41 |
B2u |
214 |
207 |
0.70 |
|
|
|
| 42 |
B3g |
3185 |
3082 |
0.00 |
|
|
|
| 43 |
B3g |
3165 |
3063 |
0.00 |
|
|
|
| 44 |
B3g |
1642 |
1588 |
0.00 |
|
|
|
| 45 |
B3g |
1483 |
1435 |
0.00 |
|
|
|
| 46 |
B3g |
1318 |
1276 |
0.00 |
|
|
|
| 47 |
B3g |
1111 |
1075 |
0.00 |
|
|
|
| 48 |
B3g |
1013 |
980 |
0.00 |
|
|
|
| 49 |
B3g |
618 |
598 |
0.00 |
|
|
|
| 50 |
B3g |
570 |
551 |
0.00 |
|
|
|
| 51 |
B3u |
945 |
915 |
7.82 |
|
|
|
| 52 |
B3u |
753 |
729 |
138.38 |
|
|
|
| 53 |
B3u |
381 |
369 |
3.17 |
|
|
|
| 54 |
B3u |
105 |
101 |
2.80 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34690.8 cm
-1
Scaled (by 0.9675) Zero Point Vibrational Energy (zpe) 33563.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/aug-cc-pVTZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.411 |
|
|
|
| 2 |
C |
0.411 |
|
|
|
| 3 |
C |
0.411 |
|
|
|
| 4 |
C |
0.411 |
|
|
|
| 5 |
C |
-0.849 |
|
|
|
| 6 |
C |
-0.308 |
|
|
|
| 7 |
C |
-0.308 |
|
|
|
| 8 |
C |
-0.849 |
|
|
|
| 9 |
C |
-0.849 |
|
|
|
| 10 |
C |
-0.308 |
|
|
|
| 11 |
C |
-0.308 |
|
|
|
| 12 |
C |
-0.849 |
|
|
|
| 13 |
H |
0.353 |
|
|
|
| 14 |
H |
0.392 |
|
|
|
| 15 |
H |
0.392 |
|
|
|
| 16 |
H |
0.353 |
|
|
|
| 17 |
H |
0.353 |
|
|
|
| 18 |
H |
0.392 |
|
|
|
| 19 |
H |
0.392 |
|
|
|
| 20 |
H |
0.353 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-75.045 |
0.000 |
0.000 |
| y |
0.000 |
-61.509 |
0.000 |
| z |
0.000 |
0.000 |
-62.071 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-13.255 |
0.000 |
0.000 |
| y |
0.000 |
7.049 |
0.000 |
| z |
0.000 |
0.000 |
6.206 |
|
| Polar |
|---|
| 3z2-r2 | 12.412 |
|---|
| x2-y2 | -13.536 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.159 |
0.000 |
0.000 |
| y |
0.000 |
20.908 |
0.000 |
| z |
0.000 |
0.000 |
34.070 |
<r2> (average value of r
2) Å
2
| <r2> |
560.188 |
| (<r2>)1/2 |
23.668 |