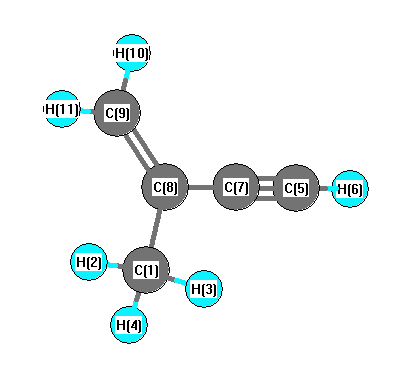
Vibrational Frequencies calculated at B3LYP/aug-cc-pVTZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3467 |
3354 |
74.63 |
|
|
|
| 2 |
A' |
3233 |
3128 |
5.85 |
|
|
|
| 3 |
A' |
3146 |
3044 |
3.24 |
|
|
|
| 4 |
A' |
3111 |
3010 |
15.46 |
|
|
|
| 5 |
A' |
3029 |
2931 |
17.17 |
|
|
|
| 6 |
A' |
2197 |
2126 |
3.61 |
|
|
|
| 7 |
A' |
1672 |
1618 |
19.23 |
|
|
|
| 8 |
A' |
1495 |
1447 |
11.83 |
|
|
|
| 9 |
A' |
1440 |
1393 |
0.06 |
|
|
|
| 10 |
A' |
1412 |
1366 |
3.79 |
|
|
|
| 11 |
A' |
1286 |
1245 |
11.98 |
|
|
|
| 12 |
A' |
1034 |
1001 |
3.00 |
|
|
|
| 13 |
A' |
967 |
936 |
0.98 |
|
|
|
| 14 |
A' |
776 |
750 |
1.20 |
|
|
|
| 15 |
A' |
677 |
655 |
40.80 |
|
|
|
| 16 |
A' |
572 |
553 |
9.73 |
|
|
|
| 17 |
A' |
398 |
385 |
0.15 |
|
|
|
| 18 |
A' |
189 |
183 |
1.80 |
|
|
|
| 19 |
A" |
3079 |
2979 |
11.52 |
|
|
|
| 20 |
A" |
1477 |
1429 |
7.51 |
|
|
|
| 21 |
A" |
1073 |
1038 |
0.18 |
|
|
|
| 22 |
A" |
942 |
912 |
41.14 |
|
|
|
| 23 |
A" |
746 |
722 |
0.03 |
|
|
|
| 24 |
A" |
657 |
635 |
42.93 |
|
|
|
| 25 |
A" |
550 |
532 |
1.33 |
|
|
|
| 26 |
A" |
272 |
263 |
9.32 |
|
|
|
| 27 |
A" |
178 |
172 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 19537.5 cm
-1
Scaled (by 0.9675) Zero Point Vibrational Energy (zpe) 18902.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.