Jump to
S1C2
Energy calculated at B3LYP/6-31G(2df,p)
| | hartrees |
|---|
| Energy at 0K | -312.317545 |
| Energy at 298.15K | -312.333034 |
| HF Energy | -312.317545 |
| Nuclear repulsion energy | 314.238701 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3107 |
2999 |
44.19 |
|
|
|
| 2 |
A1 |
3039 |
2932 |
56.20 |
|
|
|
| 3 |
A1 |
3031 |
2925 |
9.43 |
|
|
|
| 4 |
A1 |
2946 |
2843 |
85.63 |
|
|
|
| 5 |
A1 |
1538 |
1485 |
0.97 |
|
|
|
| 6 |
A1 |
1509 |
1456 |
3.84 |
|
|
|
| 7 |
A1 |
1493 |
1441 |
0.00 |
|
|
|
| 8 |
A1 |
1458 |
1406 |
1.64 |
|
|
|
| 9 |
A1 |
1412 |
1363 |
1.24 |
|
|
|
| 10 |
A1 |
1341 |
1294 |
1.98 |
|
|
|
| 11 |
A1 |
1172 |
1131 |
2.40 |
|
|
|
| 12 |
A1 |
1063 |
1026 |
6.36 |
|
|
|
| 13 |
A1 |
1003 |
968 |
16.40 |
|
|
|
| 14 |
A1 |
923 |
891 |
5.92 |
|
|
|
| 15 |
A1 |
434 |
419 |
0.42 |
|
|
|
| 16 |
A1 |
317 |
306 |
0.19 |
|
|
|
| 17 |
A1 |
104 |
100 |
0.17 |
|
|
|
| 18 |
A2 |
3098 |
2990 |
0.00 |
|
|
|
| 19 |
A2 |
3070 |
2963 |
0.00 |
|
|
|
| 20 |
A2 |
2963 |
2859 |
0.00 |
|
|
|
| 21 |
A2 |
1500 |
1448 |
0.00 |
|
|
|
| 22 |
A2 |
1314 |
1268 |
0.00 |
|
|
|
| 23 |
A2 |
1266 |
1222 |
0.00 |
|
|
|
| 24 |
A2 |
1181 |
1140 |
0.00 |
|
|
|
| 25 |
A2 |
896 |
865 |
0.00 |
|
|
|
| 26 |
A2 |
765 |
738 |
0.00 |
|
|
|
| 27 |
A2 |
232 |
224 |
0.00 |
|
|
|
| 28 |
A2 |
138 |
134 |
0.00 |
|
|
|
| 29 |
A2 |
77 |
74 |
0.00 |
|
|
|
| 30 |
B1 |
3099 |
2990 |
117.77 |
|
|
|
| 31 |
B1 |
3071 |
2963 |
9.23 |
|
|
|
| 32 |
B1 |
2962 |
2858 |
108.40 |
|
|
|
| 33 |
B1 |
1500 |
1448 |
10.22 |
|
|
|
| 34 |
B1 |
1316 |
1270 |
0.64 |
|
|
|
| 35 |
B1 |
1275 |
1230 |
3.57 |
|
|
|
| 36 |
B1 |
1205 |
1163 |
4.18 |
|
|
|
| 37 |
B1 |
908 |
876 |
2.78 |
|
|
|
| 38 |
B1 |
766 |
740 |
2.97 |
|
|
|
| 39 |
B1 |
234 |
226 |
0.26 |
|
|
|
| 40 |
B1 |
159 |
153 |
4.41 |
|
|
|
| 41 |
B1 |
56 |
54 |
0.22 |
|
|
|
| 42 |
B2 |
3107 |
2998 |
21.29 |
|
|
|
| 43 |
B2 |
3038 |
2932 |
6.28 |
|
|
|
| 44 |
B2 |
3031 |
2925 |
34.80 |
|
|
|
| 45 |
B2 |
2933 |
2830 |
15.19 |
|
|
|
| 46 |
B2 |
1519 |
1466 |
8.69 |
|
|
|
| 47 |
B2 |
1506 |
1453 |
0.00 |
|
|
|
| 48 |
B2 |
1491 |
1439 |
2.00 |
|
|
|
| 49 |
B2 |
1414 |
1365 |
10.96 |
|
|
|
| 50 |
B2 |
1405 |
1356 |
25.96 |
|
|
|
| 51 |
B2 |
1327 |
1281 |
21.78 |
|
|
|
| 52 |
B2 |
1162 |
1121 |
250.22 |
|
|
|
| 53 |
B2 |
1123 |
1084 |
0.10 |
|
|
|
| 54 |
B2 |
1048 |
1012 |
0.12 |
|
|
|
| 55 |
B2 |
897 |
865 |
1.04 |
|
|
|
| 56 |
B2 |
512 |
494 |
4.37 |
|
|
|
| 57 |
B2 |
249 |
240 |
0.57 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 42351.1 cm
-1
Scaled (by 0.965) Zero Point Vibrational Energy (zpe) 40868.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G(2df,p)
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.376 |
| C2 |
0.000 |
1.180 |
-0.399 |
| C3 |
0.000 |
-1.180 |
-0.399 |
| C4 |
0.000 |
2.382 |
0.535 |
| C5 |
0.000 |
-2.382 |
0.535 |
| C6 |
0.000 |
3.712 |
-0.222 |
| C7 |
0.000 |
-3.712 |
-0.222 |
| H8 |
0.886 |
1.207 |
-1.058 |
| H9 |
-0.886 |
1.207 |
-1.058 |
| H10 |
-0.886 |
-1.207 |
-1.058 |
| H11 |
0.886 |
-1.207 |
-1.058 |
| H12 |
0.878 |
2.315 |
1.188 |
| H13 |
-0.878 |
2.315 |
1.188 |
| H14 |
-0.878 |
-2.315 |
1.188 |
| H15 |
0.878 |
-2.315 |
1.188 |
| H16 |
0.000 |
4.559 |
0.470 |
| H17 |
-0.884 |
3.808 |
-0.862 |
| H18 |
0.884 |
3.808 |
-0.862 |
| H19 |
0.000 |
-4.559 |
0.470 |
| H20 |
0.884 |
-3.808 |
-0.862 |
| H21 |
-0.884 |
-3.808 |
-0.862 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4109 | 1.4109 | 2.3877 | 2.3877 | 3.7602 | 3.7602 | 2.0727 | 2.0727 | 2.0727 | 2.0727 | 2.6060 | 2.6060 | 2.6060 | 2.6060 | 4.5604 | 4.1006 | 4.1006 | 4.5604 | 4.1006 | 4.1006 |
C2 | 1.4109 | | 2.3591 | 1.5228 | 3.6823 | 2.5391 | 4.8952 | 1.1048 | 1.1048 | 2.6297 | 2.6297 | 2.1394 | 2.1394 | 3.9371 | 3.9371 | 3.4898 | 2.8117 | 2.8117 | 5.8044 | 5.0866 | 5.0866 | C3 | 1.4109 | 2.3591 | | 3.6823 | 1.5228 | 4.8952 | 2.5391 | 2.6297 | 2.6297 | 1.1048 | 1.1048 | 3.9371 | 3.9371 | 2.1394 | 2.1394 | 5.8044 | 5.0866 | 5.0866 | 3.4898 | 2.8117 | 2.8117 | C4 | 2.3877 | 1.5228 | 3.6823 | | 4.7648 | 1.5304 | 6.1417 | 2.1689 | 2.1689 | 4.0257 | 4.0257 | 1.0961 | 1.0961 | 4.8233 | 4.8233 | 2.1780 | 2.1834 | 2.1834 | 6.9421 | 6.4076 | 6.4076 | C5 | 2.3877 | 3.6823 | 1.5228 | 4.7648 | | 6.1417 | 1.5304 | 4.0257 | 4.0257 | 2.1689 | 2.1689 | 4.8233 | 4.8233 | 1.0961 | 1.0961 | 6.9421 | 6.4076 | 6.4076 | 2.1780 | 2.1834 | 2.1834 | C6 | 3.7602 | 2.5391 | 4.8952 | 1.5304 | 6.1417 | | 7.4249 | 2.7859 | 2.7859 | 5.0681 | 5.0681 | 2.1702 | 2.1702 | 6.2522 | 6.2522 | 1.0938 | 1.0958 | 1.0958 | 8.3008 | 7.5994 | 7.5994 | C7 | 3.7602 | 4.8952 | 2.5391 | 6.1417 | 1.5304 | 7.4249 | | 5.0681 | 5.0681 | 2.7859 | 2.7859 | 6.2522 | 6.2522 | 2.1702 | 2.1702 | 8.3008 | 7.5994 | 7.5994 | 1.0938 | 1.0958 | 1.0958 | H8 | 2.0727 | 1.1048 | 2.6297 | 2.1689 | 4.0257 | 2.7859 | 5.0681 | | 1.7725 | 2.9949 | 2.4140 | 2.5038 | 3.0631 | 4.5344 | 4.1770 | 3.7893 | 3.1523 | 2.6084 | 6.0310 | 5.0190 | 5.3220 | H9 | 2.0727 | 1.1048 | 2.6297 | 2.1689 | 4.0257 | 2.7859 | 5.0681 | 1.7725 | | 2.4140 | 2.9949 | 3.0631 | 2.5038 | 4.1770 | 4.5344 | 3.7893 | 2.6084 | 3.1523 | 6.0310 | 5.3220 | 5.0190 | H10 | 2.0727 | 2.6297 | 1.1048 | 4.0257 | 2.1689 | 5.0681 | 2.7859 | 2.9949 | 2.4140 | | 1.7725 | 4.5344 | 4.1770 | 2.5038 | 3.0631 | 6.0310 | 5.0190 | 5.3220 | 3.7893 | 3.1523 | 2.6084 | H11 | 2.0727 | 2.6297 | 1.1048 | 4.0257 | 2.1689 | 5.0681 | 2.7859 | 2.4140 | 2.9949 | 1.7725 | | 4.1770 | 4.5344 | 3.0631 | 2.5038 | 6.0310 | 5.3220 | 5.0190 | 3.7893 | 2.6084 | 3.1523 | H12 | 2.6060 | 2.1394 | 3.9371 | 1.0961 | 4.8233 | 2.1702 | 6.2522 | 2.5038 | 3.0631 | 4.5344 | 4.1770 | | 1.7566 | 4.9524 | 4.6304 | 2.5145 | 3.0881 | 2.5360 | 6.9675 | 6.4574 | 6.6935 | H13 | 2.6060 | 2.1394 | 3.9371 | 1.0961 | 4.8233 | 2.1702 | 6.2522 | 3.0631 | 2.5038 | 4.1770 | 4.5344 | 1.7566 | | 4.6304 | 4.9524 | 2.5145 | 2.5360 | 3.0881 | 6.9675 | 6.6935 | 6.4574 | H14 | 2.6060 | 3.9371 | 2.1394 | 4.8233 | 1.0961 | 6.2522 | 2.1702 | 4.5344 | 4.1770 | 2.5038 | 3.0631 | 4.9524 | 4.6304 | | 1.7566 | 6.9675 | 6.4574 | 6.6935 | 2.5145 | 3.0881 | 2.5360 | H15 | 2.6060 | 3.9371 | 2.1394 | 4.8233 | 1.0961 | 6.2522 | 2.1702 | 4.1770 | 4.5344 | 3.0631 | 2.5038 | 4.6304 | 4.9524 | 1.7566 | | 6.9675 | 6.6935 | 6.4574 | 2.5145 | 2.5360 | 3.0881 | H16 | 4.5604 | 3.4898 | 5.8044 | 2.1780 | 6.9421 | 1.0938 | 8.3008 | 3.7893 | 3.7893 | 6.0310 | 6.0310 | 2.5145 | 2.5145 | 6.9675 | 6.9675 | | 1.7669 | 1.7669 | 9.1189 | 8.5190 | 8.5190 | H17 | 4.1006 | 2.8117 | 5.0866 | 2.1834 | 6.4076 | 1.0958 | 7.5994 | 3.1523 | 2.6084 | 5.0190 | 5.3220 | 3.0881 | 2.5360 | 6.4574 | 6.6935 | 1.7669 | | 1.7677 | 8.5190 | 7.8187 | 7.6163 | H18 | 4.1006 | 2.8117 | 5.0866 | 2.1834 | 6.4076 | 1.0958 | 7.5994 | 2.6084 | 3.1523 | 5.3220 | 5.0190 | 2.5360 | 3.0881 | 6.6935 | 6.4574 | 1.7669 | 1.7677 | | 8.5190 | 7.6163 | 7.8187 | H19 | 4.5604 | 5.8044 | 3.4898 | 6.9421 | 2.1780 | 8.3008 | 1.0938 | 6.0310 | 6.0310 | 3.7893 | 3.7893 | 6.9675 | 6.9675 | 2.5145 | 2.5145 | 9.1189 | 8.5190 | 8.5190 | | 1.7669 | 1.7669 | H20 | 4.1006 | 5.0866 | 2.8117 | 6.4076 | 2.1834 | 7.5994 | 1.0958 | 5.0190 | 5.3220 | 3.1523 | 2.6084 | 6.4574 | 6.6935 | 3.0881 | 2.5360 | 8.5190 | 7.8187 | 7.6163 | 1.7669 | | 1.7677 | H21 | 4.1006 | 5.0866 | 2.8117 | 6.4076 | 2.1834 | 7.5994 | 1.0958 | 5.3220 | 5.0190 | 2.6084 | 3.1523 | 6.6935 | 6.4574 | 2.5360 | 3.0881 | 8.5190 | 7.6163 | 7.8187 | 1.7669 | 1.7677 | |
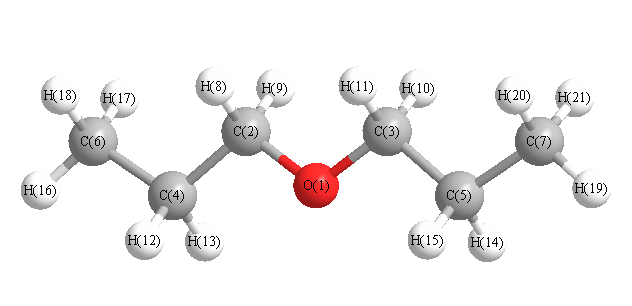 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
108.899 |
|
O1 |
C2 |
H8 |
110.369 |
| O1 |
C2 |
H9 |
110.369 |
|
O1 |
C3 |
C5 |
108.899 |
| O1 |
C3 |
H10 |
110.369 |
|
O1 |
C3 |
H11 |
110.369 |
| C2 |
O1 |
C3 |
113.451 |
|
C2 |
C4 |
C6 |
112.528 |
| C2 |
C4 |
H12 |
108.455 |
|
C2 |
C4 |
H13 |
108.455 |
| C3 |
C5 |
C7 |
112.528 |
|
C3 |
C5 |
H14 |
108.455 |
| C3 |
C5 |
H15 |
108.455 |
|
C4 |
C2 |
H8 |
110.255 |
| C4 |
C2 |
H9 |
110.255 |
|
C4 |
C6 |
H16 |
111.100 |
| C4 |
C6 |
H17 |
111.412 |
|
C4 |
C6 |
H18 |
111.412 |
| C5 |
C3 |
H10 |
110.255 |
|
C5 |
C3 |
H11 |
110.255 |
| C5 |
C7 |
H19 |
111.100 |
|
C5 |
C7 |
H20 |
111.412 |
| C5 |
C7 |
H21 |
111.412 |
|
C6 |
C4 |
H12 |
110.345 |
| C6 |
C4 |
H13 |
110.345 |
|
C7 |
C5 |
H14 |
110.345 |
| C7 |
C5 |
H15 |
110.345 |
|
H8 |
C2 |
H9 |
106.682 |
| H10 |
C3 |
H11 |
106.682 |
|
H12 |
C4 |
H13 |
106.504 |
| H14 |
C5 |
H15 |
106.504 |
|
H16 |
C6 |
H17 |
107.597 |
| H16 |
C6 |
H18 |
107.597 |
|
H17 |
C6 |
H18 |
107.526 |
| H19 |
C7 |
H20 |
107.597 |
|
H19 |
C7 |
H21 |
107.597 |
| H20 |
C7 |
H21 |
107.526 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.248 |
|
|
|
| 2 |
C |
-0.030 |
|
|
|
| 3 |
C |
-0.030 |
|
|
|
| 4 |
C |
-0.181 |
|
|
|
| 5 |
C |
-0.181 |
|
|
|
| 6 |
C |
-0.382 |
|
|
|
| 7 |
C |
-0.382 |
|
|
|
| 8 |
H |
0.079 |
|
|
|
| 9 |
H |
0.079 |
|
|
|
| 10 |
H |
0.079 |
|
|
|
| 11 |
H |
0.079 |
|
|
|
| 12 |
H |
0.106 |
|
|
|
| 13 |
H |
0.106 |
|
|
|
| 14 |
H |
0.106 |
|
|
|
| 15 |
H |
0.106 |
|
|
|
| 16 |
H |
0.116 |
|
|
|
| 17 |
H |
0.115 |
|
|
|
| 18 |
H |
0.115 |
|
|
|
| 19 |
H |
0.116 |
|
|
|
| 20 |
H |
0.115 |
|
|
|
| 21 |
H |
0.115 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-0.804 |
0.804 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-45.538 |
0.000 |
0.000 |
| y |
0.000 |
-43.684 |
0.000 |
| z |
0.000 |
0.000 |
-46.701 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-0.345 |
0.000 |
0.000 |
| y |
0.000 |
2.435 |
0.000 |
| z |
0.000 |
0.000 |
-2.090 |
|
| Polar |
|---|
| 3z2-r2 | -4.180 |
|---|
| x2-y2 | -1.854 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.298 |
0.000 |
0.000 |
| y |
0.000 |
13.474 |
0.000 |
| z |
0.000 |
0.000 |
9.607 |
<r2> (average value of r
2) Å
2
| <r2> |
435.292 |
| (<r2>)1/2 |
20.864 |
Jump to
S1C1
Energy calculated at B3LYP/6-31G(2df,p)
| | hartrees |
|---|
| Energy at 0K | -312.318310 |
| Energy at 298.15K | -312.333993 |
| HF Energy | -312.318310 |
| Nuclear repulsion energy | 324.716696 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G(2df,p)
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3125 |
3016 |
6.47 |
|
|
|
| 2 |
A |
3101 |
2992 |
57.22 |
|
|
|
| 3 |
A |
3068 |
2960 |
12.45 |
|
|
|
| 4 |
A |
3036 |
2929 |
6.09 |
|
|
|
| 5 |
A |
3033 |
2927 |
3.29 |
|
|
|
| 6 |
A |
2971 |
2867 |
9.77 |
|
|
|
| 7 |
A |
2948 |
2845 |
89.58 |
|
|
|
| 8 |
A |
1532 |
1479 |
2.19 |
|
|
|
| 9 |
A |
1511 |
1458 |
2.99 |
|
|
|
| 10 |
A |
1496 |
1443 |
5.31 |
|
|
|
| 11 |
A |
1478 |
1426 |
0.69 |
|
|
|
| 12 |
A |
1451 |
1400 |
4.56 |
|
|
|
| 13 |
A |
1406 |
1357 |
0.89 |
|
|
|
| 14 |
A |
1379 |
1331 |
1.51 |
|
|
|
| 15 |
A |
1313 |
1267 |
2.49 |
|
|
|
| 16 |
A |
1266 |
1222 |
0.10 |
|
|
|
| 17 |
A |
1182 |
1141 |
5.91 |
|
|
|
| 18 |
A |
1144 |
1104 |
9.31 |
|
|
|
| 19 |
A |
1079 |
1042 |
5.74 |
|
|
|
| 20 |
A |
954 |
921 |
6.24 |
|
|
|
| 21 |
A |
921 |
889 |
6.55 |
|
|
|
| 22 |
A |
899 |
868 |
0.50 |
|
|
|
| 23 |
A |
752 |
726 |
0.38 |
|
|
|
| 24 |
A |
480 |
463 |
0.07 |
|
|
|
| 25 |
A |
333 |
321 |
0.30 |
|
|
|
| 26 |
A |
266 |
257 |
0.64 |
|
|
|
| 27 |
A |
166 |
160 |
0.04 |
|
|
|
| 28 |
A |
112 |
108 |
0.01 |
|
|
|
| 29 |
A |
45 |
44 |
0.00 |
|
|
|
| 30 |
B |
3125 |
3015 |
49.86 |
|
|
|
| 31 |
B |
3101 |
2992 |
27.50 |
|
|
|
| 32 |
B |
3067 |
2960 |
30.81 |
|
|
|
| 33 |
B |
3036 |
2929 |
49.78 |
|
|
|
| 34 |
B |
3033 |
2926 |
63.23 |
|
|
|
| 35 |
B |
2968 |
2865 |
104.58 |
|
|
|
| 36 |
B |
2935 |
2833 |
14.92 |
|
|
|
| 37 |
B |
1511 |
1458 |
0.37 |
|
|
|
| 38 |
B |
1508 |
1456 |
11.24 |
|
|
|
| 39 |
B |
1495 |
1442 |
5.43 |
|
|
|
| 40 |
B |
1478 |
1426 |
2.15 |
|
|
|
| 41 |
B |
1411 |
1362 |
17.82 |
|
|
|
| 42 |
B |
1393 |
1344 |
29.87 |
|
|
|
| 43 |
B |
1375 |
1326 |
22.15 |
|
|
|
| 44 |
B |
1304 |
1258 |
1.95 |
|
|
|
| 45 |
B |
1277 |
1232 |
6.59 |
|
|
|
| 46 |
B |
1185 |
1143 |
7.48 |
|
|
|
| 47 |
B |
1164 |
1123 |
166.85 |
|
|
|
| 48 |
B |
1105 |
1066 |
30.44 |
|
|
|
| 49 |
B |
1025 |
989 |
30.90 |
|
|
|
| 50 |
B |
933 |
901 |
0.88 |
|
|
|
| 51 |
B |
877 |
846 |
0.16 |
|
|
|
| 52 |
B |
786 |
758 |
2.27 |
|
|
|
| 53 |
B |
496 |
478 |
1.00 |
|
|
|
| 54 |
B |
322 |
310 |
0.57 |
|
|
|
| 55 |
B |
219 |
212 |
2.13 |
|
|
|
| 56 |
B |
167 |
161 |
3.49 |
|
|
|
| 57 |
B |
75 |
73 |
1.15 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 42405.6 cm
-1
Scaled (by 0.965) Zero Point Vibrational Energy (zpe) 40921.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G(2df,p)
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| O1 |
0.000 |
0.000 |
0.123 |
| C2 |
0.023 |
1.181 |
0.896 |
| C3 |
-0.023 |
-1.181 |
0.896 |
| C4 |
0.000 |
2.384 |
-0.039 |
| C5 |
0.000 |
-2.384 |
-0.039 |
| C6 |
-1.274 |
2.464 |
-0.885 |
| C7 |
1.274 |
-2.464 |
-0.885 |
| H8 |
-0.850 |
1.208 |
1.574 |
| H9 |
0.925 |
1.203 |
1.531 |
| H10 |
0.850 |
-1.208 |
1.574 |
| H11 |
-0.925 |
-1.203 |
1.531 |
| H12 |
0.880 |
2.331 |
-0.692 |
| H13 |
0.111 |
3.291 |
0.568 |
| H14 |
-0.880 |
-2.331 |
-0.692 |
| H15 |
-0.111 |
-3.291 |
0.568 |
| H16 |
-1.245 |
3.320 |
-1.566 |
| H17 |
-1.396 |
1.555 |
-1.481 |
| H18 |
-2.162 |
2.570 |
-0.251 |
| H19 |
1.245 |
-3.320 |
-1.566 |
| H20 |
1.396 |
-1.555 |
-1.481 |
| H21 |
2.162 |
-2.570 |
-0.251 |
Atom - Atom Distances (Å)
| |
O1 |
C2 |
C3 |
C4 |
C5 |
C6 |
C7 |
H8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
H21 |
| O1 | | 1.4119 | 1.4119 | 2.3898 | 2.3898 | 2.9510 | 2.9510 | 2.0703 | 2.0702 | 2.0703 | 2.0702 | 2.6217 | 3.3224 | 2.6217 | 3.3224 | 3.9274 | 2.6340 | 3.3791 | 3.9274 | 2.6340 | 3.3791 |
C2 | 1.4119 | | 2.3633 | 1.5237 | 3.6863 | 2.5488 | 4.2455 | 1.1050 | 1.1036 | 2.6180 | 2.6436 | 2.1394 | 2.1362 | 3.9590 | 4.4859 | 3.4986 | 2.7924 | 2.8310 | 5.2741 | 3.8758 | 4.4682 | C3 | 1.4119 | 2.3633 | | 3.6863 | 1.5237 | 4.2455 | 2.5488 | 2.6180 | 2.6436 | 1.1050 | 1.1036 | 3.9590 | 4.4859 | 2.1394 | 2.1362 | 5.2741 | 3.8758 | 4.4682 | 3.4986 | 2.7924 | 2.8310 | C4 | 2.3898 | 1.5237 | 3.6863 | | 4.7685 | 1.5311 | 5.0833 | 2.1695 | 2.1714 | 4.0286 | 4.0240 | 1.0966 | 1.0968 | 4.8412 | 5.7083 | 2.1809 | 2.1706 | 2.1799 | 6.0347 | 4.4210 | 5.4096 | C5 | 2.3898 | 3.6863 | 1.5237 | 4.7685 | | 5.0833 | 1.5311 | 4.0286 | 4.0240 | 2.1695 | 2.1714 | 4.8412 | 5.7083 | 1.0966 | 1.0968 | 6.0347 | 4.4210 | 5.4096 | 2.1809 | 2.1706 | 2.1799 | C6 | 2.9510 | 2.5488 | 4.2455 | 1.5311 | 5.0833 | | 5.5470 | 2.7932 | 3.5012 | 4.9028 | 4.4053 | 2.1665 | 2.1712 | 4.8151 | 6.0478 | 1.0942 | 1.0929 | 1.0961 | 6.3450 | 4.8614 | 6.1273 | C7 | 2.9510 | 4.2455 | 2.5488 | 5.0833 | 1.5311 | 5.5470 | | 4.9028 | 4.4053 | 2.7932 | 3.5012 | 4.8151 | 6.0478 | 2.1665 | 2.1712 | 6.3450 | 4.8614 | 6.1273 | 1.0942 | 1.0929 | 1.0961 | H8 | 2.0703 | 1.1050 | 2.6180 | 2.1695 | 4.0286 | 2.7932 | 4.9028 | | 1.7749 | 2.9540 | 2.4132 | 3.0634 | 2.5039 | 4.2025 | 4.6685 | 3.8043 | 3.1220 | 2.6281 | 5.8948 | 4.6911 | 5.1647 | H9 | 2.0702 | 1.1036 | 2.6436 | 2.1714 | 4.0240 | 3.5012 | 4.4053 | 1.7749 | | 2.4132 | 3.0355 | 2.4926 | 2.4381 | 4.5488 | 4.7112 | 4.3333 | 3.8178 | 3.8170 | 5.4911 | 4.1111 | 4.3526 | H10 | 2.0703 | 2.6180 | 1.1050 | 4.0286 | 2.1695 | 4.9028 | 2.7932 | 2.9540 | 2.4132 | | 1.7749 | 4.2025 | 4.6685 | 3.0634 | 2.5039 | 5.8948 | 4.6911 | 5.1647 | 3.8043 | 3.1220 | 2.6281 | H11 | 2.0702 | 2.6436 | 1.1036 | 4.0240 | 2.1714 | 4.4053 | 3.5012 | 2.4132 | 3.0355 | 1.7749 | | 4.5488 | 4.7112 | 2.4926 | 2.4381 | 5.4911 | 4.1111 | 4.3526 | 4.3333 | 3.8178 | 3.8170 | H12 | 2.6217 | 2.1394 | 3.9590 | 1.0966 | 4.8412 | 2.1665 | 4.8151 | 3.0634 | 2.4926 | 4.2025 | 4.5488 | | 1.7603 | 4.9839 | 5.8460 | 2.5016 | 2.5303 | 3.0825 | 5.7302 | 3.9994 | 5.0854 | H13 | 3.3224 | 2.1362 | 4.4859 | 1.0968 | 5.7083 | 2.1712 | 6.0478 | 2.5039 | 2.4381 | 4.6685 | 4.7112 | 1.7603 | | 5.8460 | 6.5848 | 2.5289 | 3.0787 | 2.5211 | 7.0384 | 5.4158 | 6.2629 | H14 | 2.6217 | 3.9590 | 2.1394 | 4.8412 | 1.0966 | 4.8151 | 2.1665 | 4.2025 | 4.5488 | 3.0634 | 2.4926 | 4.9839 | 5.8460 | | 1.7603 | 5.7302 | 3.9994 | 5.0854 | 2.5016 | 2.5303 | 3.0825 | H15 | 3.3224 | 4.4859 | 2.1362 | 5.7083 | 1.0968 | 6.0478 | 2.1712 | 4.6685 | 4.7112 | 2.5039 | 2.4381 | 5.8460 | 6.5848 | 1.7603 | | 7.0384 | 5.4158 | 6.2629 | 2.5289 | 3.0787 | 2.5211 | H16 | 3.9274 | 3.4986 | 5.2741 | 2.1809 | 6.0347 | 1.0942 | 6.3450 | 3.8043 | 4.3333 | 5.8948 | 5.4911 | 2.5016 | 2.5289 | 5.7302 | 7.0384 | | 1.7729 | 1.7692 | 7.0915 | 5.5453 | 6.9302 | H17 | 2.6340 | 2.7924 | 3.8758 | 2.1706 | 4.4210 | 1.0929 | 4.8614 | 3.1220 | 3.8178 | 4.6911 | 4.1111 | 2.5303 | 3.0787 | 3.9994 | 5.4158 | 1.7729 | | 1.7685 | 5.5453 | 4.1794 | 5.5843 | H18 | 3.3791 | 2.8310 | 4.4682 | 2.1799 | 5.4096 | 1.0961 | 6.1273 | 2.6281 | 3.8170 | 5.1647 | 4.3526 | 3.0825 | 2.5211 | 5.0854 | 6.2629 | 1.7692 | 1.7685 | | 6.9302 | 5.5843 | 6.7166 | H19 | 3.9274 | 5.2741 | 3.4986 | 6.0347 | 2.1809 | 6.3450 | 1.0942 | 5.8948 | 5.4911 | 3.8043 | 4.3333 | 5.7302 | 7.0384 | 2.5016 | 2.5289 | 7.0915 | 5.5453 | 6.9302 | | 1.7729 | 1.7692 | H20 | 2.6340 | 3.8758 | 2.7924 | 4.4210 | 2.1706 | 4.8614 | 1.0929 | 4.6911 | 4.1111 | 3.1220 | 3.8178 | 3.9994 | 5.4158 | 2.5303 | 3.0787 | 5.5453 | 4.1794 | 5.5843 | 1.7729 | | 1.7685 | H21 | 3.3791 | 4.4682 | 2.8310 | 5.4096 | 2.1799 | 6.1273 | 1.0961 | 5.1647 | 4.3526 | 2.6281 | 3.8170 | 5.0854 | 6.2629 | 3.0825 | 2.5211 | 6.9302 | 5.5843 | 6.7166 | 1.7692 | 1.7685 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| O1 |
C2 |
C4 |
108.930 |
|
O1 |
C2 |
H8 |
110.093 |
| O1 |
C2 |
H9 |
110.166 |
|
O1 |
C3 |
C5 |
108.930 |
| O1 |
C3 |
H10 |
110.093 |
|
O1 |
C3 |
H11 |
110.166 |
| C2 |
O1 |
C3 |
113.630 |
|
C2 |
C4 |
C6 |
113.096 |
| C2 |
C4 |
H12 |
108.366 |
|
C2 |
C4 |
H13 |
108.102 |
| C3 |
C5 |
C7 |
113.096 |
|
C3 |
C5 |
H14 |
108.366 |
| C3 |
C5 |
H15 |
108.102 |
|
C4 |
C2 |
H8 |
110.226 |
| C4 |
C2 |
H9 |
110.455 |
|
C4 |
C6 |
H16 |
111.260 |
| C4 |
C6 |
H17 |
110.518 |
|
C4 |
C6 |
H18 |
111.068 |
| C5 |
C3 |
H10 |
110.226 |
|
C5 |
C3 |
H11 |
110.455 |
| C5 |
C7 |
H19 |
111.260 |
|
C5 |
C7 |
H20 |
110.518 |
| C5 |
C7 |
H21 |
111.068 |
|
C6 |
C4 |
H12 |
109.973 |
| C6 |
C4 |
H13 |
110.332 |
|
C7 |
C5 |
H14 |
109.973 |
| C7 |
C5 |
H15 |
110.332 |
|
H8 |
C2 |
H9 |
106.962 |
| H10 |
C3 |
H11 |
106.962 |
|
H12 |
C4 |
H13 |
106.745 |
| H14 |
C5 |
H15 |
106.745 |
|
H16 |
C6 |
H17 |
108.318 |
| H16 |
C6 |
H18 |
107.753 |
|
H17 |
C6 |
H18 |
107.790 |
| H19 |
C7 |
H20 |
108.318 |
|
H19 |
C7 |
H21 |
107.753 |
| H20 |
C7 |
H21 |
107.790 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G(2df,p)
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
O |
-0.242 |
|
|
|
| 2 |
C |
-0.034 |
|
|
|
| 3 |
C |
-0.034 |
|
|
|
| 4 |
C |
-0.187 |
|
|
|
| 5 |
C |
-0.187 |
|
|
|
| 6 |
C |
-0.364 |
|
|
|
| 7 |
C |
-0.364 |
|
|
|
| 8 |
H |
0.079 |
|
|
|
| 9 |
H |
0.081 |
|
|
|
| 10 |
H |
0.079 |
|
|
|
| 11 |
H |
0.081 |
|
|
|
| 12 |
H |
0.105 |
|
|
|
| 13 |
H |
0.093 |
|
|
|
| 14 |
H |
0.105 |
|
|
|
| 15 |
H |
0.093 |
|
|
|
| 16 |
H |
0.108 |
|
|
|
| 17 |
H |
0.132 |
|
|
|
| 18 |
H |
0.108 |
|
|
|
| 19 |
H |
0.108 |
|
|
|
| 20 |
H |
0.132 |
|
|
|
| 21 |
H |
0.108 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.866 |
0.866 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.439 |
0.358 |
0.000 |
| y |
0.358 |
-43.916 |
0.000 |
| z |
0.000 |
0.000 |
-45.513 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.725 |
0.358 |
0.000 |
| y |
0.358 |
2.061 |
0.000 |
| z |
0.000 |
0.000 |
-0.336 |
|
| Polar |
|---|
| 3z2-r2 | -0.672 |
|---|
| x2-y2 | -2.524 |
|---|
| xy | 0.358 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
9.871 |
-0.561 |
0.000 |
| y |
-0.561 |
12.187 |
0.000 |
| z |
0.000 |
0.000 |
9.998 |
<r2> (average value of r
2) Å
2
| <r2> |
344.895 |
| (<r2>)1/2 |
18.571 |