Vibrational Frequencies calculated at B3LYP/3-21G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3228 |
3114 |
8.07 |
|
|
|
| 2 |
A' |
3222 |
3109 |
8.85 |
|
|
|
| 3 |
A' |
3212 |
3100 |
14.59 |
|
|
|
| 4 |
A' |
3202 |
3090 |
6.90 |
|
|
|
| 5 |
A' |
3192 |
3080 |
0.02 |
|
|
|
| 6 |
A' |
3165 |
3054 |
10.53 |
|
|
|
| 7 |
A' |
3057 |
2950 |
1.61 |
|
|
|
| 8 |
A' |
1717 |
1657 |
86.48 |
|
|
|
| 9 |
A' |
1632 |
1574 |
15.57 |
|
|
|
| 10 |
A' |
1612 |
1556 |
8.97 |
|
|
|
| 11 |
A' |
1543 |
1489 |
0.71 |
|
|
|
| 12 |
A' |
1525 |
1472 |
14.93 |
|
|
|
| 13 |
A' |
1505 |
1452 |
16.12 |
|
|
|
| 14 |
A' |
1430 |
1380 |
38.89 |
|
|
|
| 15 |
A' |
1386 |
1338 |
5.76 |
|
|
|
| 16 |
A' |
1329 |
1282 |
1.94 |
|
|
|
| 17 |
A' |
1272 |
1227 |
167.23 |
|
|
|
| 18 |
A' |
1238 |
1194 |
30.14 |
|
|
|
| 19 |
A' |
1228 |
1185 |
14.55 |
|
|
|
| 20 |
A' |
1123 |
1083 |
7.43 |
|
|
|
| 21 |
A' |
1114 |
1075 |
1.01 |
|
|
|
| 22 |
A' |
1061 |
1024 |
7.27 |
|
|
|
| 23 |
A' |
1036 |
1000 |
0.08 |
|
|
|
| 24 |
A' |
962 |
928 |
38.09 |
|
|
|
| 25 |
A' |
750 |
723 |
0.02 |
|
|
|
| 26 |
A' |
658 |
635 |
0.75 |
|
|
|
| 27 |
A' |
590 |
569 |
21.36 |
|
|
|
| 28 |
A' |
476 |
459 |
0.48 |
|
|
|
| 29 |
A' |
373 |
360 |
1.43 |
|
|
|
| 30 |
A' |
225 |
217 |
5.11 |
|
|
|
| 31 |
A" |
3113 |
3004 |
6.59 |
|
|
|
| 32 |
A" |
1548 |
1494 |
13.09 |
|
|
|
| 33 |
A" |
1095 |
1056 |
2.60 |
|
|
|
| 34 |
A" |
1049 |
1012 |
0.73 |
|
|
|
| 35 |
A" |
1024 |
988 |
0.60 |
|
|
|
| 36 |
A" |
971 |
936 |
3.25 |
|
|
|
| 37 |
A" |
882 |
851 |
0.29 |
|
|
|
| 38 |
A" |
809 |
781 |
21.46 |
|
|
|
| 39 |
A" |
734 |
708 |
58.73 |
|
|
|
| 40 |
A" |
634 |
612 |
11.36 |
|
|
|
| 41 |
A" |
442 |
426 |
0.91 |
|
|
|
| 42 |
A" |
430 |
415 |
0.01 |
|
|
|
| 43 |
A" |
194 |
187 |
0.78 |
|
|
|
| 44 |
A" |
160 |
154 |
0.00 |
|
|
|
| 45 |
A" |
83 |
80 |
2.85 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30614.4 cm
-1
Scaled (by 0.9649) Zero Point Vibrational Energy (zpe) 29539.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
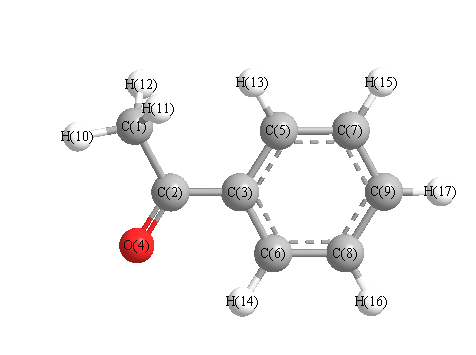
Charges from optimized geometry at B3LYP/3-21G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.633 |
|
|
|
| 2 |
C |
0.423 |
|
|
|
| 3 |
C |
-0.082 |
|
|
|
| 4 |
O |
-0.455 |
|
|
|
| 5 |
C |
-0.201 |
|
|
|
| 6 |
C |
-0.156 |
|
|
|
| 7 |
C |
-0.181 |
|
|
|
| 8 |
C |
-0.186 |
|
|
|
| 9 |
C |
-0.177 |
|
|
|
| 10 |
H |
0.227 |
|
|
|
| 11 |
H |
0.217 |
|
|
|
| 12 |
H |
0.217 |
|
|
|
| 13 |
H |
0.190 |
|
|
|
| 14 |
H |
0.217 |
|
|
|
| 15 |
H |
0.192 |
|
|
|
| 16 |
H |
0.194 |
|
|
|
| 17 |
H |
0.194 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.955 |
-2.088 |
0.000 |
2.860 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.754 |
5.551 |
0.000 |
| y |
5.551 |
-52.103 |
0.000 |
| z |
0.000 |
0.000 |
-55.662 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.129 |
5.551 |
0.000 |
| y |
5.551 |
-0.395 |
0.000 |
| z |
0.000 |
0.000 |
-5.734 |
|
| Polar |
|---|
| 3z2-r2 | -11.467 |
|---|
| x2-y2 | 4.350 |
|---|
| xy | 5.551 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.845 |
0.391 |
0.000 |
| y |
0.391 |
15.897 |
0.000 |
| z |
0.000 |
0.000 |
4.235 |
<r2> (average value of r
2) Å
2
| <r2> |
345.512 |
| (<r2>)1/2 |
18.588 |