Jump to
S1C2
Energy calculated at B3LYP/STO-3G
| | hartrees |
|---|
| Energy at 0K | -543.835451 |
| Energy at 298.15K | |
| HF Energy | -543.835451 |
| Nuclear repulsion energy | 342.394642 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/STO-3G
Geometric Data calculated at B3LYP/STO-3G
Point Group is D4h
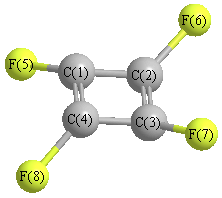
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
1.050 |
0.000 |
| C2 |
1.050 |
0.000 |
0.000 |
| C3 |
0.000 |
-1.050 |
0.000 |
| C4 |
-1.050 |
0.000 |
0.000 |
| F5 |
0.000 |
2.404 |
0.000 |
| F6 |
2.404 |
0.000 |
0.000 |
| F7 |
0.000 |
-2.404 |
0.000 |
| F8 |
-2.404 |
0.000 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.4844 | 2.0993 | 1.4844 | 1.3543 | 2.6231 | 3.4536 | 2.6231 |
C2 | 1.4844 | | 1.4844 | 2.0993 | 2.6231 | 1.3543 | 2.6231 | 3.4536 | C3 | 2.0993 | 1.4844 | | 1.4844 | 3.4536 | 2.6231 | 1.3543 | 2.6231 | C4 | 1.4844 | 2.0993 | 1.4844 | | 2.6231 | 3.4536 | 2.6231 | 1.3543 | F5 | 1.3543 | 2.6231 | 3.4536 | 2.6231 | | 3.3997 | 4.8079 | 3.3997 | F6 | 2.6231 | 1.3543 | 2.6231 | 3.4536 | 3.3997 | | 3.3997 | 4.8079 | F7 | 3.4536 | 2.6231 | 1.3543 | 2.6231 | 4.8079 | 3.3997 | | 3.3997 | F8 | 2.6231 | 3.4536 | 2.6231 | 1.3543 | 3.3997 | 4.8079 | 3.3997 | |
 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
135.000 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
135.000 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
135.000 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
135.000 |
| C3 |
C2 |
F6 |
135.000 |
|
C3 |
C4 |
F8 |
135.000 |
| C4 |
C1 |
F5 |
135.000 |
|
C4 |
C3 |
F7 |
135.000 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.141 |
|
|
|
| 2 |
C |
-0.065 |
|
|
|
| 3 |
C |
0.141 |
|
|
|
| 4 |
C |
-0.065 |
|
|
|
| 5 |
F |
0.009 |
|
|
|
| 6 |
F |
-0.085 |
|
|
|
| 7 |
F |
0.009 |
|
|
|
| 8 |
F |
-0.085 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-41.326 |
0.000 |
0.000 |
| y |
0.000 |
-31.294 |
0.000 |
| z |
0.000 |
0.000 |
-35.666 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-7.846 |
0.000 |
0.000 |
| y |
0.000 |
7.201 |
0.000 |
| z |
0.000 |
0.000 |
0.644 |
|
| Polar |
|---|
| 3z2-r2 | 1.289 |
|---|
| x2-y2 | -10.031 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.000 |
0.000 |
0.000 |
| y |
0.000 |
0.000 |
0.000 |
| z |
0.000 |
0.000 |
0.000 |
<r2> (average value of r
2) Å
2
| <r2> |
257.025 |
| (<r2>)1/2 |
16.032 |
Jump to
S1C1
Energy calculated at B3LYP/STO-3G
| | hartrees |
|---|
| Energy at 0K | -543.927655 |
| Energy at 298.15K | -543.927114 |
| HF Energy | -543.927655 |
| Nuclear repulsion energy | 341.789086 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/STO-3G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
Ag |
1939 |
1730 |
0.00 |
|
|
|
| 2 |
Ag |
1261 |
1125 |
0.00 |
|
|
|
| 3 |
Ag |
637 |
568 |
0.00 |
|
|
|
| 4 |
Ag |
257 |
229 |
0.00 |
|
|
|
| 5 |
Ag |
229 |
204 |
0.00 |
|
|
|
| 6 |
Au |
1422 |
1269 |
191.54 |
|
|
|
| 7 |
Au |
923 |
824 |
4.95 |
|
|
|
| 8 |
Au |
540 |
481 |
0.00 |
|
|
|
| 9 |
Au |
229 |
204 |
0.21 |
|
|
|
| 10 |
Au |
149 |
133 |
0.00 |
|
|
|
| 11 |
Bg |
1523 |
1359 |
0.00 |
|
|
|
| 12 |
Bg |
721 |
643 |
0.00 |
|
|
|
| 13 |
Bg |
495 |
442 |
0.00 |
|
|
|
| 14 |
Bg |
417 |
372 |
0.00 |
|
|
|
| 15 |
Bu |
1911 |
1705 |
53.91 |
|
|
|
| 16 |
Bu |
1016 |
906 |
50.74 |
|
|
|
| 17 |
Bu |
292 |
261 |
5.03 |
|
|
|
| 18 |
Bu |
202 |
180 |
0.40 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 7080.1 cm
-1
Scaled (by 0.8924) Zero Point Vibrational Energy (zpe) 6318.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/STO-3G
Point Group is C2h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.815 |
0.681 |
| C2 |
0.000 |
-0.815 |
0.681 |
| C3 |
0.000 |
-0.815 |
-0.681 |
| C4 |
0.000 |
0.815 |
-0.681 |
| F5 |
0.000 |
1.724 |
1.684 |
| F6 |
-0.000 |
-1.724 |
1.684 |
| F7 |
-0.000 |
-1.724 |
-1.684 |
| F8 |
0.000 |
1.724 |
-1.684 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
C3 |
C4 |
F5 |
F6 |
F7 |
F8 |
| C1 | | 1.6305 | 2.1242 | 1.3615 | 1.3534 | 2.7300 | 3.4696 | 2.5332 |
C2 | 1.6305 | | 1.3615 | 2.1242 | 2.7300 | 1.3534 | 2.5332 | 3.4696 | C3 | 2.1242 | 1.3615 | | 1.6305 | 3.4696 | 2.5332 | 1.3534 | 2.7300 | C4 | 1.3615 | 2.1242 | 1.6305 | | 2.5332 | 3.4696 | 2.7300 | 1.3534 | F5 | 1.3534 | 2.7300 | 3.4696 | 2.5332 | | 3.4473 | 4.8195 | 3.3680 | F6 | 2.7300 | 1.3534 | 2.5332 | 3.4696 | 3.4473 | | 3.3680 | 4.8195 | F7 | 3.4696 | 2.5332 | 1.3534 | 2.7300 | 4.8195 | 3.3680 | | 3.4473 | F8 | 2.5332 | 3.4696 | 2.7300 | 1.3534 | 3.3680 | 4.8195 | 3.4473 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
C3 |
90.000 |
|
C1 |
C2 |
F6 |
132.159 |
| C1 |
C4 |
C3 |
90.000 |
|
C1 |
C4 |
F8 |
137.841 |
| C2 |
C1 |
C4 |
90.000 |
|
C2 |
C1 |
F5 |
132.159 |
| C2 |
C3 |
C4 |
90.000 |
|
C2 |
C3 |
F7 |
137.841 |
| C3 |
C2 |
F6 |
137.841 |
|
C3 |
C4 |
F8 |
132.159 |
| C4 |
C1 |
F5 |
137.841 |
|
C4 |
C3 |
F7 |
132.159 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/STO-3G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.042 |
|
|
|
| 2 |
C |
0.042 |
|
|
|
| 3 |
C |
0.042 |
|
|
|
| 4 |
C |
0.042 |
|
|
|
| 5 |
F |
-0.042 |
|
|
|
| 6 |
F |
-0.042 |
|
|
|
| 7 |
F |
-0.042 |
|
|
|
| 8 |
F |
-0.042 |
|
|
|
Electric dipole moments
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.699 |
-0.000 |
0.000 |
| y |
-0.000 |
-36.252 |
0.000 |
| z |
0.000 |
0.000 |
-36.458 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
0.657 |
-0.000 |
0.000 |
| y |
-0.000 |
-0.174 |
0.000 |
| z |
0.000 |
0.000 |
-0.483 |
|
| Polar |
|---|
| 3z2-r2 | -0.966 |
|---|
| x2-y2 | 0.554 |
|---|
| xy | -0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
0.934 |
0.000 |
0.000 |
| y |
0.000 |
4.300 |
0.000 |
| z |
0.000 |
0.000 |
5.868 |
<r2> (average value of r
2) Å
2
| <r2> |
258.688 |
| (<r2>)1/2 |
16.084 |