Jump to
S1C2
Energy calculated at B3LYP/6-311G**
| | hartrees |
|---|
| Energy at 0K | -358.571511 |
| Energy at 298.15K | -358.576586 |
| Nuclear repulsion energy | 232.296699 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3753 |
3628 |
74.95 |
|
|
|
| 2 |
A' |
3726 |
3602 |
75.06 |
|
|
|
| 3 |
A' |
3590 |
3471 |
56.45 |
|
|
|
| 4 |
A' |
1821 |
1761 |
73.01 |
|
|
|
| 5 |
A' |
1805 |
1745 |
521.94 |
|
|
|
| 6 |
A' |
1604 |
1551 |
129.02 |
|
|
|
| 7 |
A' |
1413 |
1366 |
11.76 |
|
|
|
| 8 |
A' |
1318 |
1274 |
52.57 |
|
|
|
| 9 |
A' |
1190 |
1151 |
306.26 |
|
|
|
| 10 |
A' |
1099 |
1063 |
2.35 |
|
|
|
| 11 |
A' |
775 |
749 |
8.70 |
|
|
|
| 12 |
A' |
616 |
595 |
72.97 |
|
|
|
| 13 |
A' |
528 |
511 |
0.42 |
|
|
|
| 14 |
A' |
415 |
402 |
4.18 |
|
|
|
| 15 |
A' |
271 |
262 |
14.35 |
|
|
|
| 16 |
A" |
836 |
808 |
4.28 |
|
|
|
| 17 |
A" |
679 |
657 |
131.24 |
|
|
|
| 18 |
A" |
633 |
612 |
22.26 |
|
|
|
| 19 |
A" |
438 |
424 |
20.35 |
|
|
|
| 20 |
A" |
347 |
335 |
199.04 |
|
|
|
| 21 |
A" |
71 |
69 |
3.78 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13464.6 cm
-1
Scaled (by 0.9668) Zero Point Vibrational Energy (zpe) 13017.6 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-311G**
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.335 |
|
|
|
| 2 |
C |
0.284 |
|
|
|
| 3 |
O |
-0.320 |
|
|
|
| 4 |
O |
-0.337 |
|
|
|
| 5 |
O |
-0.280 |
|
|
|
| 6 |
N |
-0.429 |
|
|
|
| 7 |
H |
0.240 |
|
|
|
| 8 |
H |
0.250 |
|
|
|
| 9 |
H |
0.257 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
2.164 |
0.655 |
0.000 |
2.261 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.058 |
-10.098 |
0.000 |
| y |
-10.098 |
-28.550 |
0.000 |
| z |
0.000 |
0.000 |
-33.293 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.137 |
-10.098 |
0.000 |
| y |
-10.098 |
5.626 |
0.000 |
| z |
0.000 |
0.000 |
-1.489 |
|
| Polar |
|---|
| 3z2-r2 | -2.979 |
|---|
| x2-y2 | -6.509 |
|---|
| xy | -10.098 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
6.767 |
-0.213 |
0.000 |
| y |
-0.213 |
6.152 |
0.000 |
| z |
0.000 |
0.000 |
2.842 |
<r2> (average value of r
2) Å
2
| <r2> |
141.912 |
| (<r2>)1/2 |
11.913 |
Jump to
S1C1
Energy calculated at B3LYP/6-311G**
| | hartrees |
|---|
| Energy at 0K | -358.577663 |
| Energy at 298.15K | -358.582974 |
| Nuclear repulsion energy | 233.796363 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-311G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3715 |
3592 |
83.88 |
|
|
|
| 2 |
A' |
3591 |
3472 |
154.30 |
|
|
|
| 3 |
A' |
3581 |
3462 |
55.77 |
|
|
|
| 4 |
A' |
1860 |
1798 |
234.84 |
|
|
|
| 5 |
A' |
1789 |
1730 |
324.49 |
|
|
|
| 6 |
A' |
1607 |
1554 |
79.25 |
|
|
|
| 7 |
A' |
1438 |
1390 |
223.20 |
|
|
|
| 8 |
A' |
1347 |
1302 |
330.64 |
|
|
|
| 9 |
A' |
1209 |
1169 |
18.34 |
|
|
|
| 10 |
A' |
1106 |
1069 |
2.35 |
|
|
|
| 11 |
A' |
802 |
776 |
9.69 |
|
|
|
| 12 |
A' |
635 |
614 |
13.74 |
|
|
|
| 13 |
A' |
546 |
527 |
1.92 |
|
|
|
| 14 |
A' |
406 |
392 |
6.62 |
|
|
|
| 15 |
A' |
274 |
265 |
41.70 |
|
|
|
| 16 |
A" |
829 |
801 |
1.20 |
|
|
|
| 17 |
A" |
748 |
723 |
106.21 |
|
|
|
| 18 |
A" |
664 |
642 |
3.52 |
|
|
|
| 19 |
A" |
474 |
458 |
142.00 |
|
|
|
| 20 |
A" |
392 |
379 |
105.42 |
|
|
|
| 21 |
A" |
118 |
114 |
6.01 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 13564.0 cm
-1
Scaled (by 0.9668) Zero Point Vibrational Energy (zpe) 13113.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-311G**
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.010 |
-0.797 |
0.000 |
| C2 |
0.000 |
0.750 |
0.000 |
| O3 |
-1.069 |
1.341 |
0.000 |
| O4 |
1.018 |
-1.452 |
0.000 |
| O5 |
-1.224 |
-1.289 |
0.000 |
| N6 |
1.227 |
1.290 |
0.000 |
| H7 |
1.342 |
2.290 |
0.000 |
| H8 |
2.028 |
0.676 |
0.000 |
| H9 |
-1.819 |
-0.512 |
0.000 |
Atom - Atom Distances (Å)
| |
C1 |
C2 |
O3 |
O4 |
O5 |
N6 |
H7 |
H8 |
H9 |
| C1 | | 1.5476 | 2.3958 | 1.2014 | 1.3288 | 2.4161 | 3.3629 | 2.4987 | 1.8515 |
C2 | 1.5476 | | 1.2216 | 2.4261 | 2.3784 | 1.3407 | 2.0430 | 2.0293 | 2.2141 | O3 | 2.3958 | 1.2216 | | 3.4869 | 2.6350 | 2.2966 | 2.5910 | 3.1675 | 1.9997 | O4 | 1.2014 | 2.4261 | 3.4869 | | 2.2478 | 2.7501 | 3.7568 | 2.3563 | 2.9884 | O5 | 1.3288 | 2.3784 | 2.6350 | 2.2478 | | 3.5581 | 4.4044 | 3.8000 | 0.9784 | N6 | 2.4161 | 1.3407 | 2.2966 | 2.7501 | 3.5581 | | 1.0072 | 1.0087 | 3.5394 | H7 | 3.3629 | 2.0430 | 2.5910 | 3.7568 | 4.4044 | 1.0072 | | 1.7537 | 4.2246 | H8 | 2.4987 | 2.0293 | 3.1675 | 2.3563 | 3.8000 | 1.0087 | 1.7537 | | 4.0265 | H9 | 1.8515 | 2.2141 | 1.9997 | 2.9884 | 0.9784 | 3.5394 | 4.2246 | 4.0265 | |
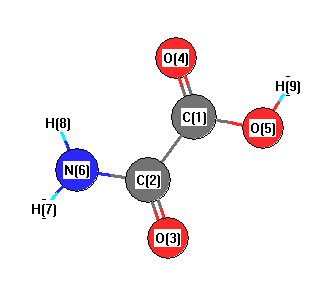 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
C2 |
O3 |
119.337 |
|
C1 |
C2 |
N6 |
113.358 |
| C1 |
O5 |
H9 |
105.734 |
|
C2 |
C1 |
O4 |
123.407 |
| C2 |
C1 |
O5 |
111.327 |
|
C2 |
N6 |
H7 |
120.288 |
| C2 |
N6 |
H8 |
118.808 |
|
O3 |
C2 |
N6 |
127.305 |
| O4 |
C1 |
O5 |
125.266 |
|
H7 |
N6 |
H8 |
120.903 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-311G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.348 |
|
|
|
| 2 |
C |
0.269 |
|
|
|
| 3 |
O |
-0.370 |
|
|
|
| 4 |
O |
-0.320 |
|
|
|
| 5 |
O |
-0.279 |
|
|
|
| 6 |
N |
-0.415 |
|
|
|
| 7 |
H |
0.243 |
|
|
|
| 8 |
H |
0.257 |
|
|
|
| 9 |
H |
0.265 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
1.527 |
2.595 |
0.000 |
3.011 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-30.453 |
7.212 |
0.000 |
| y |
7.212 |
-37.439 |
0.000 |
| z |
0.000 |
0.000 |
-33.239 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
4.886 |
7.212 |
0.000 |
| y |
7.212 |
-5.593 |
0.000 |
| z |
0.000 |
0.000 |
0.707 |
|
| Polar |
|---|
| 3z2-r2 | 1.414 |
|---|
| x2-y2 | 6.986 |
|---|
| xy | 7.212 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
7.073 |
0.040 |
0.000 |
| y |
0.040 |
5.634 |
0.000 |
| z |
0.000 |
0.000 |
2.825 |
<r2> (average value of r
2) Å
2
| <r2> |
139.599 |
| (<r2>)1/2 |
11.815 |