Jump to
S1C2
Energy calculated at B3LYP/TZVP
| | hartrees |
|---|
| Energy at 0K | -135.228549 |
| Energy at 298.15K | -135.236689 |
| HF Energy | -135.228549 |
| Nuclear repulsion energy | 82.662931 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/TZVP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3483 |
3362 |
0.79 |
|
|
|
| 2 |
A' |
3080 |
2974 |
52.05 |
|
|
|
| 3 |
A' |
3031 |
2927 |
33.49 |
|
|
|
| 4 |
A' |
3016 |
2912 |
24.24 |
|
|
|
| 5 |
A' |
1667 |
1610 |
26.34 |
|
|
|
| 6 |
A' |
1507 |
1455 |
3.17 |
|
|
|
| 7 |
A' |
1488 |
1437 |
0.25 |
|
|
|
| 8 |
A' |
1414 |
1365 |
8.10 |
|
|
|
| 9 |
A' |
1389 |
1341 |
14.09 |
|
|
|
| 10 |
A' |
1143 |
1104 |
10.17 |
|
|
|
| 11 |
A' |
1055 |
1019 |
27.35 |
|
|
|
| 12 |
A' |
889 |
858 |
27.51 |
|
|
|
| 13 |
A' |
831 |
803 |
169.86 |
|
|
|
| 14 |
A' |
403 |
389 |
7.53 |
|
|
|
| 15 |
A" |
3562 |
3439 |
0.02 |
|
|
|
| 16 |
A" |
3088 |
2981 |
66.15 |
|
|
|
| 17 |
A" |
3056 |
2950 |
3.80 |
|
|
|
| 18 |
A" |
1497 |
1445 |
7.82 |
|
|
|
| 19 |
A" |
1392 |
1343 |
0.06 |
|
|
|
| 20 |
A" |
1273 |
1229 |
0.02 |
|
|
|
| 21 |
A" |
1010 |
975 |
0.89 |
|
|
|
| 22 |
A" |
783 |
756 |
0.75 |
|
|
|
| 23 |
A" |
288 |
278 |
34.97 |
|
|
|
| 24 |
A" |
252 |
243 |
16.44 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20297.8 cm
-1
Scaled (by 0.9654) Zero Point Vibrational Energy (zpe) 19595.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/TZVP
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-1.308 |
-0.088 |
0.000 |
| C2 |
0.000 |
0.572 |
0.000 |
| C3 |
1.218 |
-0.355 |
0.000 |
| H4 |
2.152 |
0.215 |
0.000 |
| H5 |
1.220 |
-0.999 |
0.883 |
| H6 |
1.220 |
-0.999 |
-0.883 |
| H7 |
0.039 |
1.229 |
-0.873 |
| H8 |
0.039 |
1.229 |
0.873 |
| H9 |
-1.413 |
-0.681 |
0.816 |
| H10 |
-1.413 |
-0.681 |
-0.816 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
C3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.4646 | 2.5403 | 3.4733 | 2.8288 | 2.8288 | 2.0757 | 2.0757 | 1.0143 | 1.0143 |
C2 | 1.4646 | | 1.5308 | 2.1818 | 2.1767 | 2.1767 | 1.0935 | 1.0935 | 2.0572 | 2.0572 | C3 | 2.5403 | 1.5308 | | 1.0939 | 1.0936 | 1.0936 | 2.1594 | 2.1594 | 2.7741 | 2.7741 | H4 | 3.4733 | 2.1818 | 1.0939 | | 1.7675 | 1.7675 | 2.5018 | 2.5018 | 3.7656 | 3.7656 | H5 | 2.8288 | 2.1767 | 1.0936 | 1.7675 | | 1.7670 | 3.0737 | 2.5224 | 2.6530 | 3.1497 | H6 | 2.8288 | 2.1767 | 1.0936 | 1.7675 | 1.7670 | | 2.5224 | 3.0737 | 3.1497 | 2.6530 | H7 | 2.0757 | 1.0935 | 2.1594 | 2.5018 | 3.0737 | 2.5224 | | 1.7461 | 2.9340 | 2.4000 | H8 | 2.0757 | 1.0935 | 2.1594 | 2.5018 | 2.5224 | 3.0737 | 1.7461 | | 2.4000 | 2.9340 | H9 | 1.0143 | 2.0572 | 2.7741 | 3.7656 | 2.6530 | 3.1497 | 2.9340 | 2.4000 | | 1.6312 | H10 | 1.0143 | 2.0572 | 2.7741 | 3.7656 | 3.1497 | 2.6530 | 2.4000 | 2.9340 | 1.6312 | |
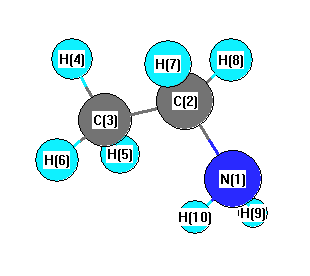 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
C3 |
115.990 |
|
N1 |
C2 |
H7 |
107.588 |
| N1 |
C2 |
H8 |
107.588 |
|
C2 |
N1 |
H9 |
110.860 |
| C2 |
N1 |
H10 |
110.860 |
|
C2 |
C3 |
H4 |
111.368 |
| C2 |
C3 |
H5 |
110.978 |
|
C2 |
C3 |
H6 |
110.978 |
| C3 |
C2 |
H7 |
109.616 |
|
C3 |
C2 |
H8 |
109.616 |
| H4 |
C3 |
H5 |
107.794 |
|
H4 |
C3 |
H6 |
107.794 |
| H5 |
C3 |
H6 |
107.769 |
|
H7 |
C2 |
H8 |
105.958 |
| H9 |
N1 |
H10 |
107.043 |
|
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/TZVP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.417 |
|
|
|
| 2 |
C |
-0.168 |
|
|
|
| 3 |
C |
-0.337 |
|
|
|
| 4 |
H |
0.108 |
|
|
|
| 5 |
H |
0.100 |
|
|
|
| 6 |
H |
0.100 |
|
|
|
| 7 |
H |
0.120 |
|
|
|
| 8 |
H |
0.120 |
|
|
|
| 9 |
H |
0.187 |
|
|
|
| 10 |
H |
0.187 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.959 |
-0.964 |
0.000 |
1.359 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-24.373 |
2.770 |
0.000 |
| y |
2.770 |
-20.743 |
0.000 |
| z |
0.000 |
0.000 |
-19.336 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-4.334 |
2.770 |
0.000 |
| y |
2.770 |
1.112 |
0.000 |
| z |
0.000 |
0.000 |
3.222 |
|
| Polar |
|---|
| 3z2-r2 | 6.445 |
|---|
| x2-y2 | -3.630 |
|---|
| xy | 2.770 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.589 |
0.093 |
0.000 |
| y |
0.093 |
5.003 |
0.000 |
| z |
0.000 |
0.000 |
4.877 |
<r2> (average value of r
2) Å
2
| <r2> |
59.084 |
| (<r2>)1/2 |
7.687 |
Jump to
S1C1
Energy calculated at B3LYP/TZVP
| | hartrees |
|---|
| Energy at 0K | -135.228377 |
| Energy at 298.15K | |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/TZVP
Geometric Data calculated at B3LYP/TZVP
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability