Jump to
S1C2
Energy calculated at B3LYP/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -151.283354 |
| Energy at 298.15K | -151.291299 |
| HF Energy | -151.283354 |
| Counterpoise corrected energy | |
| CP Energy at 298.15K | |
| Counterpoise optimized geometry corrected energy | |
| CP opt. Energy at 298.15K | |
| Nuclear repulsion energy | 83.118812 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3490 |
3359 |
0.95 |
|
|
|
| 2 |
A1 |
3049 |
2935 |
36.73 |
|
|
|
| 3 |
A1 |
1665 |
1602 |
41.25 |
|
|
|
| 4 |
A1 |
1490 |
1434 |
0.15 |
|
|
|
| 5 |
A1 |
1082 |
1042 |
37.63 |
|
|
|
| 6 |
A1 |
833 |
802 |
3.57 |
|
|
|
| 7 |
A1 |
456 |
439 |
2.43 |
|
|
|
| 8 |
A2 |
3572 |
3438 |
0.00 |
|
|
|
| 9 |
A2 |
1398 |
1345 |
0.00 |
|
|
|
| 10 |
A2 |
1072 |
1032 |
0.00 |
|
|
|
| 11 |
A2 |
253 |
243 |
0.00 |
|
|
|
| 12 |
B1 |
3570 |
3437 |
0.69 |
|
|
|
| 13 |
B1 |
3087 |
2972 |
26.42 |
|
|
|
| 14 |
B1 |
1365 |
1314 |
0.12 |
|
|
|
| 15 |
B1 |
847 |
816 |
0.12 |
|
|
|
| 16 |
B1 |
383 |
369 |
80.11 |
|
|
|
| 17 |
B2 |
3490 |
3360 |
1.25 |
|
|
|
| 18 |
B2 |
1653 |
1591 |
5.31 |
|
|
|
| 19 |
B2 |
1393 |
1341 |
16.72 |
|
|
|
| 20 |
B2 |
1059 |
1019 |
80.26 |
|
|
|
| 21 |
B2 |
764 |
736 |
422.19 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17985.5 cm
-1
Scaled (by 0.9626) Zero Point Vibrational Energy (zpe) 17312.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/Def2TZVPP
Point Group is C2v
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.539 |
| N2 |
0.000 |
1.283 |
-0.181 |
| N3 |
0.000 |
-1.283 |
-0.181 |
| H4 |
0.879 |
0.000 |
1.182 |
| H5 |
-0.879 |
0.000 |
1.182 |
| H6 |
0.818 |
1.372 |
-0.768 |
| H7 |
-0.818 |
1.372 |
-0.768 |
| H8 |
-0.818 |
-1.372 |
-0.768 |
| H9 |
0.818 |
-1.372 |
-0.768 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
N3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
| C1 | | 1.4710 | 1.4710 | 1.0889 | 1.0889 | 2.0643 | 2.0643 | 2.0643 | 2.0643 |
N2 | 1.4710 | | 2.5652 | 2.0675 | 2.0675 | 1.0108 | 1.0108 | 2.8396 | 2.8396 | N3 | 1.4710 | 2.5652 | | 2.0675 | 2.0675 | 2.8396 | 2.8396 | 1.0108 | 1.0108 | H4 | 1.0889 | 2.0675 | 2.0675 | | 1.7589 | 2.3847 | 2.9265 | 2.9265 | 2.3847 | H5 | 1.0889 | 2.0675 | 2.0675 | 1.7589 | | 2.9265 | 2.3847 | 2.3847 | 2.9265 | H6 | 2.0643 | 1.0108 | 2.8396 | 2.3847 | 2.9265 | | 1.6362 | 3.1956 | 2.7449 | H7 | 2.0643 | 1.0108 | 2.8396 | 2.9265 | 2.3847 | 1.6362 | | 2.7449 | 3.1956 | H8 | 2.0643 | 2.8396 | 1.0108 | 2.9265 | 2.3847 | 3.1956 | 2.7449 | | 1.6362 | H9 | 2.0643 | 2.8396 | 1.0108 | 2.3847 | 2.9265 | 2.7449 | 3.1956 | 1.6362 | |
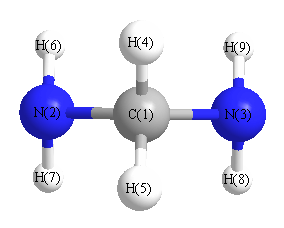 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
H6 |
111.209 |
|
C1 |
N2 |
H7 |
111.209 |
| C1 |
N3 |
H8 |
111.209 |
|
C1 |
N3 |
H9 |
111.209 |
| N2 |
C1 |
N3 |
121.372 |
|
N2 |
C1 |
H4 |
106.780 |
| N2 |
C1 |
H5 |
106.780 |
|
N3 |
C1 |
H4 |
106.780 |
| N3 |
C1 |
H5 |
106.780 |
|
H4 |
C1 |
H5 |
107.733 |
| H6 |
N2 |
H7 |
108.056 |
|
H8 |
N3 |
H9 |
108.056 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.019 |
|
|
|
| 2 |
N |
-0.319 |
|
|
|
| 3 |
N |
-0.319 |
|
|
|
| 4 |
H |
0.090 |
|
|
|
| 5 |
H |
0.090 |
|
|
|
| 6 |
H |
0.110 |
|
|
|
| 7 |
H |
0.110 |
|
|
|
| 8 |
H |
0.110 |
|
|
|
| 9 |
H |
0.110 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
-1.789 |
1.789 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-17.056 |
0.000 |
0.000 |
| y |
0.000 |
-26.082 |
0.000 |
| z |
0.000 |
0.000 |
-18.662 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
5.316 |
0.000 |
0.000 |
| y |
0.000 |
-8.222 |
0.000 |
| z |
0.000 |
0.000 |
2.907 |
|
| Polar |
|---|
| 3z2-r2 | 5.814 |
|---|
| x2-y2 | 9.025 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.518 |
0.000 |
0.000 |
| y |
0.000 |
5.183 |
0.000 |
| z |
0.000 |
0.000 |
4.622 |
<r2> (average value of r
2) Å
2
| <r2> |
54.133 |
| (<r2>)1/2 |
7.357 |
Jump to
S1C1
Energy calculated at B3LYP/Def2TZVPP
| | hartrees |
|---|
| Energy at 0K | -151.281436 |
| Energy at 298.15K | -151.289375 |
| HF Energy | -151.281436 |
| Counterpoise corrected energy | |
| CP Energy at 298.15K | |
| Counterpoise optimized geometry corrected energy | |
| CP opt. Energy at 298.15K | |
| Nuclear repulsion energy | 83.591904 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3586 |
3452 |
0.68 |
|
|
|
| 2 |
A |
3498 |
3368 |
0.36 |
|
|
|
| 3 |
A |
2946 |
2836 |
107.27 |
|
|
|
| 4 |
A |
1641 |
1580 |
1.59 |
|
|
|
| 5 |
A |
1539 |
1481 |
0.69 |
|
|
|
| 6 |
A |
1362 |
1311 |
2.82 |
|
|
|
| 7 |
A |
1211 |
1166 |
4.27 |
|
|
|
| 8 |
A |
965 |
929 |
4.96 |
|
|
|
| 9 |
A |
846 |
815 |
165.97 |
|
|
|
| 10 |
A |
489 |
470 |
0.77 |
|
|
|
| 11 |
A |
292 |
282 |
20.09 |
|
|
|
| 12 |
B |
3587 |
3453 |
2.27 |
|
|
|
| 13 |
B |
3499 |
3368 |
0.08 |
|
|
|
| 14 |
B |
2959 |
2849 |
87.07 |
|
|
|
| 15 |
B |
1638 |
1576 |
67.07 |
|
|
|
| 16 |
B |
1455 |
1401 |
31.94 |
|
|
|
| 17 |
B |
1251 |
1204 |
12.75 |
|
|
|
| 18 |
B |
1126 |
1083 |
32.21 |
|
|
|
| 19 |
B |
977 |
941 |
15.71 |
|
|
|
| 20 |
B |
815 |
784 |
148.12 |
|
|
|
| 21 |
B |
266 |
256 |
68.97 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 17973.6 cm
-1
Scaled (by 0.9626) Zero Point Vibrational Energy (zpe) 17301.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/Def2TZVPP
Point Group is C2
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| C1 |
0.000 |
0.000 |
0.545 |
| N2 |
0.000 |
1.177 |
-0.311 |
| N3 |
0.000 |
-1.177 |
-0.311 |
| H4 |
0.880 |
-0.079 |
1.200 |
| H5 |
-0.880 |
0.079 |
1.200 |
| H6 |
-0.038 |
2.033 |
0.227 |
| H7 |
0.836 |
1.194 |
-0.883 |
| H8 |
0.038 |
-2.033 |
0.227 |
| H9 |
-0.836 |
-1.194 |
-0.883 |
Atom - Atom Distances (Å)
| |
C1 |
N2 |
N3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
| C1 | | 1.4559 | 1.4559 | 1.0992 | 1.0992 | 2.0582 | 2.0409 | 2.0582 | 2.0409 |
N2 | 1.4559 | | 2.3547 | 2.1530 | 2.0644 | 1.0118 | 1.0131 | 3.2556 | 2.5786 | N3 | 1.4559 | 2.3547 | | 2.0644 | 2.1530 | 3.2556 | 2.5786 | 1.0118 | 1.0131 | H4 | 1.0992 | 2.1530 | 2.0644 | | 1.7662 | 2.5001 | 2.4418 | 2.3389 | 2.9196 | H5 | 1.0992 | 2.0644 | 2.1530 | 1.7662 | | 2.3389 | 2.9196 | 2.5001 | 2.4418 | H6 | 2.0582 | 1.0118 | 3.2556 | 2.5001 | 2.3389 | | 1.6437 | 4.0670 | 3.5048 | H7 | 2.0409 | 1.0131 | 2.5786 | 2.4418 | 2.9196 | 1.6437 | | 3.5048 | 2.9150 | H8 | 2.0582 | 3.2556 | 1.0118 | 2.3389 | 2.5001 | 4.0670 | 3.5048 | | 1.6437 | H9 | 2.0409 | 2.5786 | 1.0131 | 2.9196 | 2.4418 | 3.5048 | 2.9150 | 1.6437 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| C1 |
N2 |
H6 |
111.776 |
|
C1 |
N2 |
H7 |
110.212 |
| C1 |
N3 |
H8 |
111.776 |
|
C1 |
N3 |
H9 |
110.212 |
| N2 |
C1 |
N3 |
107.937 |
|
N2 |
C1 |
H4 |
114.118 |
| N2 |
C1 |
H5 |
106.971 |
|
N3 |
C1 |
H4 |
106.971 |
| N3 |
C1 |
H5 |
114.118 |
|
H4 |
C1 |
H5 |
106.913 |
| H6 |
N2 |
H7 |
108.537 |
|
H8 |
N3 |
H9 |
108.537 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
0.031 |
|
|
|
| 2 |
N |
-0.344 |
|
|
|
| 3 |
N |
-0.344 |
|
|
|
| 4 |
H |
0.077 |
|
|
|
| 5 |
H |
0.077 |
|
|
|
| 6 |
H |
0.117 |
|
|
|
| 7 |
H |
0.135 |
|
|
|
| 8 |
H |
0.117 |
|
|
|
| 9 |
H |
0.135 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.405 |
1.405 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-20.150 |
2.688 |
0.000 |
| y |
2.688 |
-16.949 |
0.000 |
| z |
0.000 |
0.000 |
-20.883 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-1.234 |
2.688 |
0.000 |
| y |
2.688 |
3.568 |
0.000 |
| z |
0.000 |
0.000 |
-2.334 |
|
| Polar |
|---|
| 3z2-r2 | -4.667 |
|---|
| x2-y2 | -3.201 |
|---|
| xy | 2.688 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
4.260 |
0.040 |
0.000 |
| y |
0.040 |
5.477 |
0.000 |
| z |
0.000 |
0.000 |
4.694 |
<r2> (average value of r
2) Å
2
| <r2> |
53.214 |
| (<r2>)1/2 |
7.295 |