Vibrational Frequencies calculated at B3LYP/Def2TZVPP
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3195 |
3076 |
4.94 |
|
|
|
| 2 |
A' |
3189 |
3069 |
17.89 |
|
|
|
| 3 |
A' |
3164 |
3046 |
11.52 |
|
|
|
| 4 |
A' |
3159 |
3041 |
2.23 |
|
|
|
| 5 |
A' |
3017 |
2905 |
1.60 |
|
|
|
| 6 |
A' |
1731 |
1666 |
291.58 |
|
|
|
| 7 |
A' |
1693 |
1630 |
21.49 |
|
|
|
| 8 |
A' |
1607 |
1547 |
31.45 |
|
|
|
| 9 |
A' |
1452 |
1397 |
7.02 |
|
|
|
| 10 |
A' |
1411 |
1358 |
4.55 |
|
|
|
| 11 |
A' |
1402 |
1350 |
29.10 |
|
|
|
| 12 |
A' |
1337 |
1287 |
7.55 |
|
|
|
| 13 |
A' |
1250 |
1203 |
19.38 |
|
|
|
| 14 |
A' |
1196 |
1151 |
5.34 |
|
|
|
| 15 |
A' |
1164 |
1121 |
16.43 |
|
|
|
| 16 |
A' |
1001 |
963 |
13.93 |
|
|
|
| 17 |
A' |
960 |
924 |
1.36 |
|
|
|
| 18 |
A' |
949 |
914 |
5.78 |
|
|
|
| 19 |
A' |
747 |
719 |
8.63 |
|
|
|
| 20 |
A' |
581 |
560 |
1.02 |
|
|
|
| 21 |
A' |
496 |
478 |
12.12 |
|
|
|
| 22 |
A' |
458 |
440 |
8.47 |
|
|
|
| 23 |
A" |
3035 |
2922 |
0.93 |
|
|
|
| 24 |
A" |
1194 |
1149 |
4.36 |
|
|
|
| 25 |
A" |
1033 |
994 |
0.38 |
|
|
|
| 26 |
A" |
1007 |
969 |
1.08 |
|
|
|
| 27 |
A" |
948 |
913 |
1.48 |
|
|
|
| 28 |
A" |
817 |
786 |
4.58 |
|
|
|
| 29 |
A" |
728 |
701 |
59.85 |
|
|
|
| 30 |
A" |
541 |
521 |
8.04 |
|
|
|
| 31 |
A" |
448 |
431 |
0.08 |
|
|
|
| 32 |
A" |
273 |
263 |
0.01 |
|
|
|
| 33 |
A" |
55 |
53 |
6.52 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 22618.4 cm
-1
Scaled (by 0.9626) Zero Point Vibrational Energy (zpe) 21772.5 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/Def2TZVPP
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
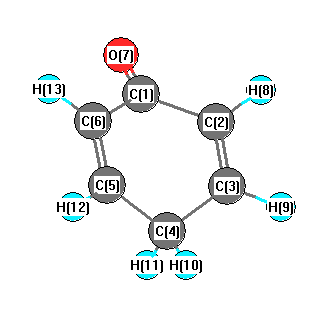
| 1 |
C |
0.151 |
|
|
|
| 2 |
C |
-0.157 |
|
|
|
| 3 |
C |
-0.081 |
|
|
|
| 4 |
C |
-0.105 |
|
|
|
| 5 |
C |
-0.171 |
|
|
|
| 6 |
C |
-0.037 |
|
|
|
| 7 |
O |
-0.310 |
|
|
|
| 8 |
H |
0.128 |
|
|
|
| 9 |
H |
0.135 |
|
|
|
| 10 |
H |
0.128 |
|
|
|
| 11 |
H |
0.127 |
|
|
|
| 12 |
H |
0.096 |
|
|
|
| 13 |
H |
0.096 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.330 |
-3.680 |
0.000 |
3.695 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-35.457 |
-0.354 |
0.000 |
| y |
-0.354 |
-47.939 |
0.000 |
| z |
0.000 |
0.000 |
-42.288 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
9.657 |
-0.354 |
0.000 |
| y |
-0.354 |
-9.067 |
0.000 |
| z |
0.000 |
0.000 |
-0.590 |
|
| Polar |
|---|
| 3z2-r2 | -1.179 |
|---|
| x2-y2 | 12.482 |
|---|
| xy | -0.354 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
11.661 |
-0.023 |
0.000 |
| y |
-0.023 |
13.304 |
0.000 |
| z |
0.000 |
0.000 |
6.357 |
<r2> (average value of r
2) Å
2
| <r2> |
186.379 |
| (<r2>)1/2 |
13.652 |