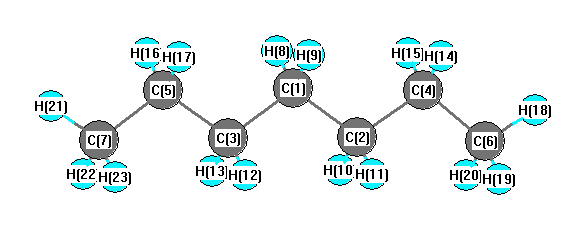
Vibrational Frequencies calculated at B3LYP/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3099 |
2988 |
55.53 |
|
|
|
| 2 |
A1 |
3029 |
2921 |
38.89 |
|
|
|
| 3 |
A1 |
3021 |
2913 |
157.28 |
|
|
|
| 4 |
A1 |
3009 |
2901 |
11.81 |
|
|
|
| 5 |
A1 |
3003 |
2896 |
0.23 |
|
|
|
| 6 |
A1 |
1519 |
1465 |
10.20 |
|
|
|
| 7 |
A1 |
1507 |
1453 |
1.04 |
|
|
|
| 8 |
A1 |
1495 |
1442 |
0.12 |
|
|
|
| 9 |
A1 |
1492 |
1438 |
0.02 |
|
|
|
| 10 |
A1 |
1418 |
1367 |
3.11 |
|
|
|
| 11 |
A1 |
1402 |
1352 |
0.22 |
|
|
|
| 12 |
A1 |
1320 |
1273 |
0.04 |
|
|
|
| 13 |
A1 |
1156 |
1114 |
0.75 |
|
|
|
| 14 |
A1 |
1067 |
1029 |
0.70 |
|
|
|
| 15 |
A1 |
996 |
960 |
0.00 |
|
|
|
| 16 |
A1 |
918 |
885 |
1.08 |
|
|
|
| 17 |
A1 |
417 |
402 |
0.02 |
|
|
|
| 18 |
A1 |
301 |
290 |
0.00 |
|
|
|
| 19 |
A1 |
98 |
95 |
0.00 |
|
|
|
| 20 |
A2 |
3095 |
2984 |
0.00 |
|
|
|
| 21 |
A2 |
3054 |
2945 |
0.00 |
|
|
|
| 22 |
A2 |
3029 |
2921 |
0.00 |
|
|
|
| 23 |
A2 |
1504 |
1450 |
0.00 |
|
|
|
| 24 |
A2 |
1334 |
1286 |
0.00 |
|
|
|
| 25 |
A2 |
1326 |
1278 |
0.00 |
|
|
|
| 26 |
A2 |
1243 |
1198 |
0.00 |
|
|
|
| 27 |
A2 |
1029 |
992 |
0.00 |
|
|
|
| 28 |
A2 |
851 |
820 |
0.00 |
|
|
|
| 29 |
A2 |
741 |
714 |
0.00 |
|
|
|
| 30 |
A2 |
242 |
233 |
0.00 |
|
|
|
| 31 |
A2 |
146 |
141 |
0.00 |
|
|
|
| 32 |
A2 |
81 |
79 |
0.00 |
|
|
|
| 33 |
B1 |
3095 |
2984 |
142.78 |
|
|
|
| 34 |
B1 |
3065 |
2955 |
116.57 |
|
|
|
| 35 |
B1 |
3041 |
2932 |
0.12 |
|
|
|
| 36 |
B1 |
3024 |
2915 |
0.08 |
|
|
|
| 37 |
B1 |
1504 |
1450 |
16.63 |
|
|
|
| 38 |
B1 |
1340 |
1292 |
1.03 |
|
|
|
| 39 |
B1 |
1294 |
1248 |
0.27 |
|
|
|
| 40 |
B1 |
1206 |
1163 |
0.02 |
|
|
|
| 41 |
B1 |
942 |
909 |
0.60 |
|
|
|
| 42 |
B1 |
777 |
749 |
1.09 |
|
|
|
| 43 |
B1 |
734 |
708 |
4.06 |
|
|
|
| 44 |
B1 |
244 |
236 |
0.00 |
|
|
|
| 45 |
B1 |
149 |
144 |
0.00 |
|
|
|
| 46 |
B1 |
68 |
66 |
0.00 |
|
|
|
| 47 |
B2 |
3099 |
2988 |
37.10 |
|
|
|
| 48 |
B2 |
3029 |
2920 |
57.66 |
|
|
|
| 49 |
B2 |
3017 |
2909 |
0.01 |
|
|
|
| 50 |
B2 |
3003 |
2895 |
0.03 |
|
|
|
| 51 |
B2 |
1514 |
1460 |
1.30 |
|
|
|
| 52 |
B2 |
1500 |
1446 |
1.81 |
|
|
|
| 53 |
B2 |
1491 |
1438 |
0.33 |
|
|
|
| 54 |
B2 |
1419 |
1368 |
1.74 |
|
|
|
| 55 |
B2 |
1406 |
1356 |
1.19 |
|
|
|
| 56 |
B2 |
1371 |
1322 |
2.00 |
|
|
|
| 57 |
B2 |
1260 |
1215 |
2.59 |
|
|
|
| 58 |
B2 |
1089 |
1050 |
3.27 |
|
|
|
| 59 |
B2 |
1066 |
1028 |
0.21 |
|
|
|
| 60 |
B2 |
1039 |
1001 |
0.22 |
|
|
|
| 61 |
B2 |
881 |
849 |
1.90 |
|
|
|
| 62 |
B2 |
481 |
464 |
0.11 |
|
|
|
| 63 |
B2 |
244 |
235 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 47665.7 cm
-1
Scaled (by 0.9642) Zero Point Vibrational Energy (zpe) 45959.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.179 |
|
|
|
| 2 |
C |
-0.250 |
|
|
|
| 3 |
C |
-0.250 |
|
|
|
| 4 |
C |
-0.102 |
|
|
|
| 5 |
C |
-0.102 |
|
|
|
| 6 |
C |
-0.664 |
|
|
|
| 7 |
C |
-0.664 |
|
|
|
| 8 |
H |
0.134 |
|
|
|
| 9 |
H |
0.134 |
|
|
|
| 10 |
H |
0.134 |
|
|
|
| 11 |
H |
0.134 |
|
|
|
| 12 |
H |
0.134 |
|
|
|
| 13 |
H |
0.134 |
|
|
|
| 14 |
H |
0.136 |
|
|
|
| 15 |
H |
0.136 |
|
|
|
| 16 |
H |
0.136 |
|
|
|
| 17 |
H |
0.136 |
|
|
|
| 18 |
H |
0.143 |
|
|
|
| 19 |
H |
0.144 |
|
|
|
| 20 |
H |
0.144 |
|
|
|
| 21 |
H |
0.143 |
|
|
|
| 22 |
H |
0.144 |
|
|
|
| 23 |
H |
0.144 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.083 |
0.083 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-48.251 |
0.000 |
0.000 |
| y |
0.000 |
-50.498 |
0.000 |
| z |
0.000 |
0.000 |
-49.959 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.978 |
0.000 |
0.000 |
| y |
0.000 |
-1.393 |
0.000 |
| z |
0.000 |
0.000 |
-0.584 |
|
| Polar |
|---|
| 3z2-r2 | -1.169 |
|---|
| x2-y2 | 2.247 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.871 |
0.000 |
0.000 |
| y |
0.000 |
15.376 |
0.000 |
| z |
0.000 |
0.000 |
11.800 |
<r2> (average value of r
2) Å
2
| <r2> |
0.000 |
| (<r2>)1/2 |
0.000 |