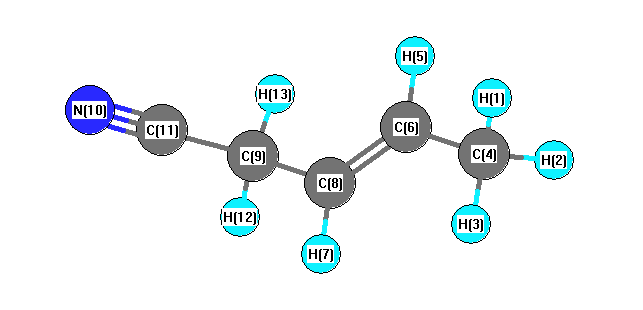
Vibrational Frequencies calculated at B3LYP/6-31+G**
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3163 |
3050 |
15.46 |
|
|
|
| 2 |
A |
3136 |
3024 |
26.82 |
|
|
|
| 3 |
A |
3115 |
3004 |
5.78 |
|
|
|
| 4 |
A |
3087 |
2977 |
2.23 |
|
|
|
| 5 |
A |
3081 |
2971 |
15.93 |
|
|
|
| 6 |
A |
3034 |
2926 |
6.56 |
|
|
|
| 7 |
A |
3030 |
2921 |
26.67 |
|
|
|
| 8 |
A |
2353 |
2269 |
13.68 |
|
|
|
| 9 |
A |
1740 |
1678 |
1.99 |
|
|
|
| 10 |
A |
1494 |
1441 |
13.21 |
|
|
|
| 11 |
A |
1484 |
1431 |
8.68 |
|
|
|
| 12 |
A |
1464 |
1411 |
10.08 |
|
|
|
| 13 |
A |
1421 |
1370 |
2.68 |
|
|
|
| 14 |
A |
1359 |
1310 |
1.74 |
|
|
|
| 15 |
A |
1334 |
1286 |
0.35 |
|
|
|
| 16 |
A |
1304 |
1257 |
7.21 |
|
|
|
| 17 |
A |
1226 |
1182 |
0.12 |
|
|
|
| 18 |
A |
1135 |
1094 |
0.65 |
|
|
|
| 19 |
A |
1073 |
1034 |
4.71 |
|
|
|
| 20 |
A |
1070 |
1032 |
0.89 |
|
|
|
| 21 |
A |
996 |
961 |
44.12 |
|
|
|
| 22 |
A |
956 |
922 |
10.21 |
|
|
|
| 23 |
A |
936 |
902 |
2.91 |
|
|
|
| 24 |
A |
900 |
868 |
1.38 |
|
|
|
| 25 |
A |
770 |
742 |
0.64 |
|
|
|
| 26 |
A |
566 |
546 |
0.96 |
|
|
|
| 27 |
A |
449 |
433 |
0.39 |
|
|
|
| 28 |
A |
374 |
361 |
0.56 |
|
|
|
| 29 |
A |
292 |
282 |
1.29 |
|
|
|
| 30 |
A |
264 |
255 |
5.66 |
|
|
|
| 31 |
A |
206 |
199 |
1.31 |
|
|
|
| 32 |
A |
136 |
131 |
4.08 |
|
|
|
| 33 |
A |
71 |
69 |
2.28 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 23509.7 cm
-1
Scaled (by 0.9642) Zero Point Vibrational Energy (zpe) 22668.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31+G**
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
H |
0.164 |
|
|
0.096 |
| 2 |
H |
0.154 |
|
|
0.084 |
| 3 |
H |
0.160 |
|
|
0.083 |
| 4 |
C |
-0.732 |
|
|
-0.239 |
| 5 |
H |
0.123 |
|
|
0.126 |
| 6 |
C |
0.161 |
|
|
-0.069 |
| 7 |
H |
0.135 |
|
|
0.165 |
| 8 |
C |
-0.062 |
|
|
-0.219 |
| 9 |
C |
-0.134 |
|
|
-0.125 |
| 10 |
N |
-0.498 |
|
|
-0.491 |
| 11 |
C |
0.154 |
|
|
0.381 |
| 12 |
H |
0.180 |
|
|
0.100 |
| 13 |
H |
0.195 |
|
|
0.107 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
3.792 |
2.340 |
0.297 |
4.466 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
3.778 |
2.355 |
0.304 |
4.462 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-47.099 |
-6.603 |
-1.861 |
| y |
-6.603 |
-37.633 |
-1.327 |
| z |
-1.861 |
-1.327 |
-35.707 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-10.429 |
-6.603 |
-1.861 |
| y |
-6.603 |
3.770 |
-1.327 |
| z |
-1.861 |
-1.327 |
6.660 |
|
| Polar |
|---|
| 3z2-r2 | 13.319 |
|---|
| x2-y2 | -9.466 |
|---|
| xy | -6.603 |
|---|
| xz | -1.861 |
|---|
| yz | -1.327 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.988 |
0.654 |
-0.783 |
| y |
0.654 |
7.918 |
-0.153 |
| z |
-0.783 |
-0.153 |
7.508 |
<r2> (average value of r
2) Å
2
| <r2> |
237.367 |
| (<r2>)1/2 |
15.407 |