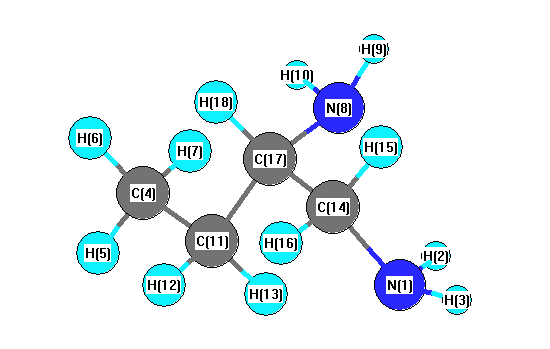
Vibrational Frequencies calculated at B3LYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3537 |
3431 |
0.89 |
|
|
|
| 2 |
A |
3535 |
3429 |
0.30 |
|
|
|
| 3 |
A |
3459 |
3355 |
2.26 |
|
|
|
| 4 |
A |
3456 |
3352 |
5.72 |
|
|
|
| 5 |
A |
3103 |
3010 |
42.07 |
|
|
|
| 6 |
A |
3094 |
3001 |
38.56 |
|
|
|
| 7 |
A |
3076 |
2983 |
12.78 |
|
|
|
| 8 |
A |
3038 |
2947 |
59.13 |
|
|
|
| 9 |
A |
3032 |
2941 |
4.74 |
|
|
|
| 10 |
A |
3018 |
2928 |
32.97 |
|
|
|
| 11 |
A |
2915 |
2827 |
131.32 |
|
|
|
| 12 |
A |
2894 |
2807 |
73.14 |
|
|
|
| 13 |
A |
1638 |
1589 |
28.43 |
|
|
|
| 14 |
A |
1619 |
1571 |
35.05 |
|
|
|
| 15 |
A |
1486 |
1442 |
4.16 |
|
|
|
| 16 |
A |
1479 |
1435 |
5.90 |
|
|
|
| 17 |
A |
1472 |
1427 |
4.20 |
|
|
|
| 18 |
A |
1456 |
1413 |
1.29 |
|
|
|
| 19 |
A |
1423 |
1381 |
1.28 |
|
|
|
| 20 |
A |
1418 |
1376 |
11.10 |
|
|
|
| 21 |
A |
1398 |
1356 |
1.84 |
|
|
|
| 22 |
A |
1375 |
1334 |
12.81 |
|
|
|
| 23 |
A |
1336 |
1296 |
2.55 |
|
|
|
| 24 |
A |
1295 |
1257 |
0.59 |
|
|
|
| 25 |
A |
1283 |
1244 |
1.85 |
|
|
|
| 26 |
A |
1257 |
1219 |
1.12 |
|
|
|
| 27 |
A |
1190 |
1155 |
4.10 |
|
|
|
| 28 |
A |
1149 |
1115 |
6.30 |
|
|
|
| 29 |
A |
1105 |
1072 |
1.36 |
|
|
|
| 30 |
A |
1073 |
1041 |
4.19 |
|
|
|
| 31 |
A |
1062 |
1030 |
1.78 |
|
|
|
| 32 |
A |
1009 |
979 |
1.26 |
|
|
|
| 33 |
A |
978 |
948 |
26.19 |
|
|
|
| 34 |
A |
927 |
900 |
34.66 |
|
|
|
| 35 |
A |
921 |
893 |
40.99 |
|
|
|
| 36 |
A |
871 |
844 |
117.09 |
|
|
|
| 37 |
A |
819 |
794 |
14.23 |
|
|
|
| 38 |
A |
768 |
745 |
1.00 |
|
|
|
| 39 |
A |
619 |
600 |
16.08 |
|
|
|
| 40 |
A |
447 |
434 |
5.08 |
|
|
|
| 41 |
A |
383 |
372 |
4.76 |
|
|
|
| 42 |
A |
340 |
330 |
31.19 |
|
|
|
| 43 |
A |
281 |
273 |
26.32 |
|
|
|
| 44 |
A |
268 |
260 |
27.27 |
|
|
|
| 45 |
A |
239 |
232 |
10.05 |
|
|
|
| 46 |
A |
219 |
212 |
4.30 |
|
|
|
| 47 |
A |
147 |
143 |
5.69 |
|
|
|
| 48 |
A |
82 |
80 |
1.26 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 36493.9 cm
-1
Scaled (by 0.97) Zero Point Vibrational Energy (zpe) 35399.1 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.169 |
|
|
|
| 2 |
H |
0.071 |
|
|
|
| 3 |
H |
0.065 |
|
|
|
| 4 |
C |
-0.020 |
|
|
|
| 5 |
H |
0.017 |
|
|
|
| 6 |
H |
0.012 |
|
|
|
| 7 |
H |
0.017 |
|
|
|
| 8 |
N |
-0.147 |
|
|
|
| 9 |
H |
0.070 |
|
|
|
| 10 |
H |
0.065 |
|
|
|
| 11 |
C |
0.027 |
|
|
|
| 12 |
H |
-0.002 |
|
|
|
| 13 |
H |
0.012 |
|
|
|
| 14 |
C |
0.089 |
|
|
|
| 15 |
H |
-0.024 |
|
|
|
| 16 |
H |
0.003 |
|
|
|
| 17 |
C |
-0.053 |
|
|
|
| 18 |
H |
-0.032 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.116 |
1.359 |
1.059 |
1.727 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-36.044 |
0.285 |
0.570 |
| y |
0.285 |
-38.548 |
-0.339 |
| z |
0.570 |
-0.339 |
-40.987 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.723 |
0.285 |
0.570 |
| y |
0.285 |
-0.032 |
-0.339 |
| z |
0.570 |
-0.339 |
-3.691 |
|
| Polar |
|---|
| 3z2-r2 | -7.382 |
|---|
| x2-y2 | 2.503 |
|---|
| xy | 0.285 |
|---|
| xz | 0.570 |
|---|
| yz | -0.339 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.306 |
-0.000 |
0.136 |
| y |
-0.000 |
8.709 |
-0.043 |
| z |
0.136 |
-0.043 |
8.322 |
<r2> (average value of r
2) Å
2
| <r2> |
206.879 |
| (<r2>)1/2 |
14.383 |