Jump to
S1C2
Energy calculated at B3LYP/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -135.176877 |
| Energy at 298.15K | -135.185065 |
| Nuclear repulsion energy | 82.397546 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/cc-pVDZ
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3438 |
3334 |
2.86 |
|
|
|
| 2 |
A' |
3086 |
2993 |
47.22 |
|
|
|
| 3 |
A' |
3021 |
2930 |
36.24 |
|
|
|
| 4 |
A' |
3011 |
2921 |
29.41 |
|
|
|
| 5 |
A' |
1651 |
1601 |
14.28 |
|
|
|
| 6 |
A' |
1479 |
1435 |
1.10 |
|
|
|
| 7 |
A' |
1458 |
1414 |
0.07 |
|
|
|
| 8 |
A' |
1393 |
1351 |
8.84 |
|
|
|
| 9 |
A' |
1368 |
1327 |
17.25 |
|
|
|
| 10 |
A' |
1152 |
1118 |
8.24 |
|
|
|
| 11 |
A' |
1068 |
1036 |
31.43 |
|
|
|
| 12 |
A' |
906 |
879 |
89.42 |
|
|
|
| 13 |
A' |
874 |
848 |
64.72 |
|
|
|
| 14 |
A' |
401 |
389 |
5.32 |
|
|
|
| 15 |
A" |
3510 |
3404 |
0.94 |
|
|
|
| 16 |
A" |
3088 |
2995 |
60.39 |
|
|
|
| 17 |
A" |
3051 |
2960 |
9.93 |
|
|
|
| 18 |
A" |
1465 |
1421 |
7.03 |
|
|
|
| 19 |
A" |
1385 |
1344 |
0.00 |
|
|
|
| 20 |
A" |
1263 |
1225 |
0.12 |
|
|
|
| 21 |
A" |
1007 |
977 |
1.13 |
|
|
|
| 22 |
A" |
780 |
757 |
1.18 |
|
|
|
| 23 |
A" |
313 |
304 |
34.26 |
|
|
|
| 24 |
A" |
259 |
251 |
8.63 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 20212.6 cm
-1
Scaled (by 0.97) Zero Point Vibrational Energy (zpe) 19606.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/cc-pVDZ
Point Group is Cs
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
-1.314 |
-0.075 |
0.000 |
| C2 |
0.000 |
0.574 |
0.000 |
| C3 |
1.213 |
-0.362 |
0.000 |
| H4 |
2.163 |
0.201 |
0.000 |
| H5 |
1.211 |
-1.015 |
0.891 |
| H6 |
1.211 |
-1.015 |
-0.891 |
| H7 |
0.046 |
1.239 |
-0.881 |
| H8 |
0.046 |
1.239 |
0.881 |
| H9 |
-1.381 |
-0.697 |
0.811 |
| H10 |
-1.381 |
-0.697 |
-0.811 |
Atom - Atom Distances (Å)
| |
N1 |
C2 |
C3 |
H4 |
H5 |
H6 |
H7 |
H8 |
H9 |
H10 |
| N1 | | 1.4654 | 2.5432 | 3.4874 | 2.8369 | 2.8369 | 2.0862 | 2.0862 | 1.0240 | 1.0240 |
C2 | 1.4654 | | 1.5327 | 2.1949 | 2.1873 | 2.1873 | 1.1047 | 1.1047 | 2.0443 | 2.0443 | C3 | 2.5432 | 1.5327 | | 1.1039 | 1.1044 | 1.1044 | 2.1686 | 2.1686 | 2.7382 | 2.7382 | H4 | 3.4874 | 2.1949 | 1.1039 | | 1.7828 | 1.7828 | 2.5169 | 2.5169 | 3.7442 | 3.7442 | H5 | 2.8369 | 2.1873 | 1.1044 | 1.7828 | | 1.7820 | 3.0944 | 2.5369 | 2.6117 | 3.1163 | H6 | 2.8369 | 2.1873 | 1.1044 | 1.7828 | 1.7820 | | 2.5369 | 3.0944 | 3.1163 | 2.6117 | H7 | 2.0862 | 1.1047 | 2.1686 | 2.5169 | 3.0944 | 2.5369 | | 1.7620 | 2.9404 | 2.4058 | H8 | 2.0862 | 1.1047 | 2.1686 | 2.5169 | 2.5369 | 3.0944 | 1.7620 | | 2.4058 | 2.9404 | H9 | 1.0240 | 2.0443 | 2.7382 | 3.7442 | 2.6117 | 3.1163 | 2.9404 | 2.4058 | | 1.6218 | H10 | 1.0240 | 2.0443 | 2.7382 | 3.7442 | 3.1163 | 2.6117 | 2.4058 | 2.9404 | 1.6218 | |
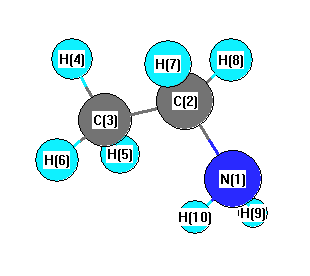 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C2 |
C3 |
116.031 |
|
N1 |
C2 |
H7 |
107.706 |
| N1 |
C2 |
H8 |
107.706 |
|
C2 |
N1 |
H9 |
109.126 |
| C2 |
N1 |
H10 |
109.126 |
|
C2 |
C3 |
H4 |
111.669 |
| C2 |
C3 |
H5 |
111.040 |
|
C2 |
C3 |
H6 |
111.040 |
| C3 |
C2 |
H7 |
109.552 |
|
C3 |
C2 |
H8 |
109.552 |
| H4 |
C3 |
H5 |
107.669 |
|
H4 |
C3 |
H6 |
107.669 |
| H5 |
C3 |
H6 |
107.567 |
|
H7 |
C2 |
H8 |
105.792 |
| H9 |
N1 |
H10 |
104.727 |
|
Electronic energy levels
Electronic state
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/cc-pVDZ
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.180 |
|
|
|
| 2 |
C |
0.014 |
|
|
|
| 3 |
C |
-0.036 |
|
|
|
| 4 |
H |
0.012 |
|
|
|
| 5 |
H |
0.012 |
|
|
|
| 6 |
H |
0.012 |
|
|
|
| 7 |
H |
0.017 |
|
|
|
| 8 |
H |
0.017 |
|
|
|
| 9 |
H |
0.067 |
|
|
|
| 10 |
H |
0.067 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.772 |
-0.932 |
0.000 |
1.211 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.294 |
-0.003 |
0.013 |
| y |
-0.003 |
4.653 |
-0.138 |
| z |
0.013 |
-0.138 |
4.261 |
<r2> (average value of r
2) Å
2
| <r2> |
58.888 |
| (<r2>)1/2 |
7.674 |
Jump to
S1C1
Energy calculated at B3LYP/cc-pVDZ
| | hartrees |
|---|
| Energy at 0K | -135.176039 |
| Energy at 298.15K | |
| Nuclear repulsion energy | |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/cc-pVDZ
Geometric Data calculated at B3LYP/cc-pVDZ
Point Group is Cs
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability