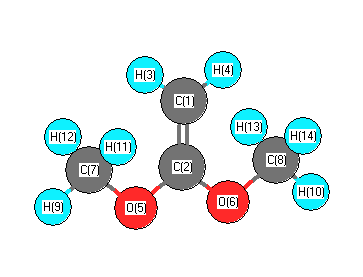
Vibrational Frequencies calculated at B3LYP/6-311G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3201 |
3094 |
0.05 |
|
|
|
| 2 |
A1 |
3147 |
3041 |
29.71 |
|
|
|
| 3 |
A1 |
3018 |
2917 |
9.10 |
|
|
|
| 4 |
A1 |
1691 |
1634 |
257.77 |
|
|
|
| 5 |
A1 |
1531 |
1479 |
6.24 |
|
|
|
| 6 |
A1 |
1504 |
1453 |
11.39 |
|
|
|
| 7 |
A1 |
1437 |
1389 |
15.52 |
|
|
|
| 8 |
A1 |
1241 |
1199 |
3.96 |
|
|
|
| 9 |
A1 |
1173 |
1133 |
0.00 |
|
|
|
| 10 |
A1 |
891 |
861 |
42.48 |
|
|
|
| 11 |
A1 |
503 |
486 |
0.61 |
|
|
|
| 12 |
A1 |
269 |
260 |
4.77 |
|
|
|
| 13 |
A2 |
3077 |
2973 |
0.00 |
|
|
|
| 14 |
A2 |
1510 |
1459 |
0.00 |
|
|
|
| 15 |
A2 |
1187 |
1147 |
0.00 |
|
|
|
| 16 |
A2 |
678 |
655 |
0.00 |
|
|
|
| 17 |
A2 |
252 |
243 |
0.00 |
|
|
|
| 18 |
A2 |
133 |
128 |
0.00 |
|
|
|
| 19 |
B1 |
3077 |
2974 |
92.88 |
|
|
|
| 20 |
B1 |
1509 |
1459 |
18.14 |
|
|
|
| 21 |
B1 |
1191 |
1151 |
1.76 |
|
|
|
| 22 |
B1 |
723 |
699 |
96.96 |
|
|
|
| 23 |
B1 |
680 |
657 |
3.60 |
|
|
|
| 24 |
B1 |
272 |
263 |
0.12 |
|
|
|
| 25 |
B1 |
101 |
97 |
9.38 |
|
|
|
| 26 |
B2 |
3279 |
3169 |
7.91 |
|
|
|
| 27 |
B2 |
3146 |
3040 |
19.29 |
|
|
|
| 28 |
B2 |
3018 |
2916 |
72.55 |
|
|
|
| 29 |
B2 |
1526 |
1475 |
17.42 |
|
|
|
| 30 |
B2 |
1504 |
1454 |
38.08 |
|
|
|
| 31 |
B2 |
1342 |
1297 |
494.70 |
|
|
|
| 32 |
B2 |
1229 |
1188 |
19.83 |
|
|
|
| 33 |
B2 |
1068 |
1032 |
123.73 |
|
|
|
| 34 |
B2 |
945 |
913 |
0.38 |
|
|
|
| 35 |
B2 |
565 |
546 |
0.32 |
|
|
|
| 36 |
B2 |
381 |
368 |
7.95 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 25998.5 cm
-1
Scaled (by 0.9663) Zero Point Vibrational Energy (zpe) 25122.3 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-311G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.662 |
|
|
-0.904 |
| 2 |
C |
0.455 |
|
|
0.720 |
| 3 |
H |
0.212 |
|
|
0.254 |
| 4 |
H |
0.212 |
|
|
0.254 |
| 5 |
O |
-0.299 |
|
|
-0.386 |
| 6 |
O |
-0.299 |
|
|
-0.386 |
| 7 |
C |
-0.458 |
|
|
0.062 |
| 8 |
C |
-0.458 |
|
|
0.062 |
| 9 |
H |
0.227 |
|
|
0.086 |
| 10 |
H |
0.227 |
|
|
0.086 |
| 11 |
H |
0.211 |
|
|
0.037 |
| 12 |
H |
0.211 |
|
|
0.039 |
| 13 |
H |
0.211 |
|
|
0.039 |
| 14 |
H |
0.211 |
|
|
0.037 |
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
1.142 |
1.142 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
0.015 |
0.000 |
1.183 |
1.183 |
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-38.754 |
0.000 |
0.000 |
| y |
0.000 |
-26.514 |
0.000 |
| z |
0.000 |
0.000 |
-39.605 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-5.694 |
0.000 |
0.000 |
| y |
0.000 |
12.665 |
0.000 |
| z |
0.000 |
0.000 |
-6.971 |
|
| Polar |
|---|
| 3z2-r2 | -13.943 |
|---|
| x2-y2 | -12.240 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.600 |
0.000 |
0.000 |
| y |
0.000 |
9.490 |
0.000 |
| z |
0.000 |
0.000 |
8.000 |
<r2> (average value of r
2) Å
2
| <r2> |
187.754 |
| (<r2>)1/2 |
13.702 |