Jump to
S1C2
Energy calculated at B3LYP/6-31G
| | hartrees |
|---|
| Energy at 0K | -345.220481 |
| Energy at 298.15K | -345.237633 |
| Nuclear repulsion energy | 420.446113 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1' |
3067 |
2950 |
0.00 |
|
|
|
| 2 |
A1' |
1535 |
1476 |
0.00 |
|
|
|
| 3 |
A1' |
1357 |
1305 |
0.00 |
|
|
|
| 4 |
A1' |
942 |
906 |
0.00 |
|
|
|
| 5 |
A1' |
793 |
763 |
0.00 |
|
|
|
| 6 |
A1' |
605 |
582 |
0.00 |
|
|
|
| 7 |
A1" |
3092 |
2974 |
0.00 |
|
|
|
| 8 |
A1" |
1283 |
1234 |
0.00 |
|
|
|
| 9 |
A1" |
1041 |
1001 |
0.00 |
|
|
|
| 10 |
A1" |
118 |
114 |
0.00 |
|
|
|
| 11 |
A2' |
3113 |
2995 |
0.00 |
|
|
|
| 12 |
A2' |
1207 |
1162 |
0.00 |
|
|
|
| 13 |
A2' |
825 |
794 |
0.00 |
|
|
|
| 14 |
A2" |
3052 |
2936 |
122.34 |
|
|
|
| 15 |
A2" |
1531 |
1473 |
5.72 |
|
|
|
| 16 |
A2" |
1398 |
1345 |
1.74 |
|
|
|
| 17 |
A2" |
993 |
955 |
19.23 |
|
|
|
| 18 |
A2" |
709 |
682 |
69.04 |
|
|
|
| 19 |
E' |
3123 |
3004 |
90.57 |
|
|
|
| 19 |
E' |
3123 |
3004 |
90.55 |
|
|
|
| 20 |
E' |
3058 |
2941 |
105.43 |
|
|
|
| 20 |
E' |
3058 |
2941 |
105.47 |
|
|
|
| 21 |
E' |
1528 |
1470 |
13.22 |
|
|
|
| 21 |
E' |
1528 |
1470 |
13.23 |
|
|
|
| 22 |
E' |
1364 |
1312 |
5.59 |
|
|
|
| 22 |
E' |
1364 |
1312 |
5.59 |
|
|
|
| 23 |
E' |
1338 |
1287 |
0.16 |
|
|
|
| 23 |
E' |
1338 |
1287 |
0.16 |
|
|
|
| 24 |
E' |
1056 |
1016 |
33.33 |
|
|
|
| 24 |
E' |
1056 |
1016 |
33.34 |
|
|
|
| 25 |
E' |
892 |
858 |
2.79 |
|
|
|
| 25 |
E' |
892 |
858 |
2.79 |
|
|
|
| 26 |
E' |
841 |
809 |
7.50 |
|
|
|
| 26 |
E' |
841 |
809 |
7.50 |
|
|
|
| 27 |
E' |
426 |
410 |
0.03 |
|
|
|
| 27 |
E' |
426 |
410 |
0.03 |
|
|
|
| 28 |
E" |
3096 |
2979 |
0.00 |
|
|
|
| 28 |
E" |
3096 |
2979 |
0.00 |
|
|
|
| 29 |
E" |
3049 |
2933 |
0.00 |
|
|
|
| 29 |
E" |
3049 |
2933 |
0.00 |
|
|
|
| 30 |
E" |
1521 |
1463 |
0.00 |
|
|
|
| 30 |
E" |
1521 |
1463 |
0.00 |
|
|
|
| 31 |
E" |
1367 |
1315 |
0.00 |
|
|
|
| 31 |
E" |
1367 |
1315 |
0.00 |
|
|
|
| 32 |
E" |
1343 |
1292 |
0.00 |
|
|
|
| 32 |
E" |
1343 |
1292 |
0.00 |
|
|
|
| 33 |
E" |
1221 |
1174 |
0.00 |
|
|
|
| 33 |
E" |
1221 |
1174 |
0.00 |
|
|
|
| 34 |
E" |
1023 |
984 |
0.00 |
|
|
|
| 34 |
E" |
1023 |
984 |
0.00 |
|
|
|
| 35 |
E" |
587 |
565 |
0.00 |
|
|
|
| 35 |
E" |
587 |
565 |
0.00 |
|
|
|
| 36 |
E" |
337 |
324 |
0.00 |
|
|
|
| 36 |
E" |
337 |
324 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40498.2 cm
-1
Scaled (by 0.962) Zero Point Vibrational Energy (zpe) 38959.2 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G
Point Group is D3h
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.282 |
| N2 |
0.000 |
0.000 |
-1.282 |
| C3 |
0.000 |
1.406 |
0.785 |
| C4 |
1.217 |
-0.703 |
0.785 |
| C5 |
-1.217 |
-0.703 |
0.785 |
| C6 |
0.000 |
1.406 |
-0.785 |
| C7 |
-1.217 |
-0.703 |
-0.785 |
| C8 |
1.217 |
-0.703 |
-0.785 |
| H9 |
0.885 |
1.908 |
1.191 |
| H10 |
-0.885 |
1.908 |
1.191 |
| H11 |
1.210 |
-1.720 |
1.191 |
| H12 |
2.095 |
-0.187 |
1.191 |
| H13 |
-2.095 |
-0.187 |
1.191 |
| H14 |
-1.210 |
-1.720 |
1.191 |
| H15 |
-0.885 |
1.908 |
-1.191 |
| H16 |
0.885 |
1.908 |
-1.191 |
| H17 |
-1.210 |
-1.720 |
-1.191 |
| H18 |
-2.095 |
-0.187 |
-1.191 |
| H19 |
2.095 |
-0.187 |
-1.191 |
| H20 |
1.210 |
-1.720 |
-1.191 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5639 | 1.4908 | 1.4908 | 1.4908 | 2.4999 | 2.4999 | 2.4999 | 2.1049 | 2.1049 | 2.1049 | 2.1049 | 2.1049 | 2.1049 | 3.2465 | 3.2465 | 3.2465 | 3.2465 | 3.2465 | 3.2465 |
N2 | 2.5639 | | 2.4999 | 2.4999 | 2.4999 | 1.4908 | 1.4908 | 1.4908 | 3.2465 | 3.2465 | 3.2465 | 3.2465 | 3.2465 | 3.2465 | 2.1049 | 2.1049 | 2.1049 | 2.1049 | 2.1049 | 2.1049 | C3 | 1.4908 | 2.4999 | | 2.4346 | 2.4346 | 1.5707 | 2.8973 | 2.8973 | 1.0955 | 1.0955 | 3.3763 | 2.6627 | 2.6627 | 3.3763 | 2.2232 | 2.2232 | 3.8913 | 3.2913 | 3.2913 | 3.8913 | C4 | 1.4908 | 2.4999 | 2.4346 | | 2.4346 | 2.8973 | 2.8973 | 1.5707 | 2.6627 | 3.3763 | 1.0955 | 1.0955 | 3.3763 | 2.6627 | 3.8913 | 3.2913 | 3.2913 | 3.8913 | 2.2232 | 2.2232 | C5 | 1.4908 | 2.4999 | 2.4346 | 2.4346 | | 2.8973 | 1.5707 | 2.8973 | 3.3763 | 2.6627 | 2.6627 | 3.3763 | 1.0955 | 1.0955 | 3.2913 | 3.8913 | 2.2232 | 2.2232 | 3.8913 | 3.2913 | C6 | 2.4999 | 1.4908 | 1.5707 | 2.8973 | 2.8973 | | 2.4346 | 2.4346 | 2.2232 | 2.2232 | 3.8913 | 3.2913 | 3.2913 | 3.8913 | 1.0955 | 1.0955 | 3.3763 | 2.6627 | 2.6627 | 3.3763 | C7 | 2.4999 | 1.4908 | 2.8973 | 2.8973 | 1.5707 | 2.4346 | | 2.4346 | 3.8913 | 3.2913 | 3.2913 | 3.8913 | 2.2232 | 2.2232 | 2.6627 | 3.3763 | 1.0955 | 1.0955 | 3.3763 | 2.6627 | C8 | 2.4999 | 1.4908 | 2.8973 | 1.5707 | 2.8973 | 2.4346 | 2.4346 | | 3.2913 | 3.8913 | 2.2232 | 2.2232 | 3.8913 | 3.2913 | 3.3763 | 2.6627 | 2.6627 | 3.3763 | 1.0955 | 1.0955 | H9 | 2.1049 | 3.2465 | 1.0955 | 2.6627 | 3.3763 | 2.2232 | 3.8913 | 3.2913 | | 1.7700 | 3.6425 | 2.4192 | 3.6425 | 4.1892 | 2.9683 | 2.3828 | 4.8195 | 4.3526 | 3.3956 | 4.3526 | H10 | 2.1049 | 3.2465 | 1.0955 | 3.3763 | 2.6627 | 2.2232 | 3.2913 | 3.8913 | 1.7700 | | 4.1892 | 3.6425 | 2.4192 | 3.6425 | 2.3828 | 2.9683 | 4.3526 | 3.3956 | 4.3526 | 4.8195 | H11 | 2.1049 | 3.2465 | 3.3763 | 1.0955 | 2.6627 | 3.8913 | 3.2913 | 2.2232 | 3.6425 | 4.1892 | | 1.7700 | 3.6425 | 2.4192 | 4.8195 | 4.3526 | 3.3956 | 4.3526 | 2.9683 | 2.3828 | H12 | 2.1049 | 3.2465 | 2.6627 | 1.0955 | 3.3763 | 3.2913 | 3.8913 | 2.2232 | 2.4192 | 3.6425 | 1.7700 | | 4.1892 | 3.6425 | 4.3526 | 3.3956 | 4.3526 | 4.8195 | 2.3828 | 2.9683 | H13 | 2.1049 | 3.2465 | 2.6627 | 3.3763 | 1.0955 | 3.2913 | 2.2232 | 3.8913 | 3.6425 | 2.4192 | 3.6425 | 4.1892 | | 1.7700 | 3.3956 | 4.3526 | 2.9683 | 2.3828 | 4.8195 | 4.3526 | H14 | 2.1049 | 3.2465 | 3.3763 | 2.6627 | 1.0955 | 3.8913 | 2.2232 | 3.2913 | 4.1892 | 3.6425 | 2.4192 | 3.6425 | 1.7700 | | 4.3526 | 4.8195 | 2.3828 | 2.9683 | 4.3526 | 3.3956 | H15 | 3.2465 | 2.1049 | 2.2232 | 3.8913 | 3.2913 | 1.0955 | 2.6627 | 3.3763 | 2.9683 | 2.3828 | 4.8195 | 4.3526 | 3.3956 | 4.3526 | | 1.7700 | 3.6425 | 2.4192 | 3.6425 | 4.1892 | H16 | 3.2465 | 2.1049 | 2.2232 | 3.2913 | 3.8913 | 1.0955 | 3.3763 | 2.6627 | 2.3828 | 2.9683 | 4.3526 | 3.3956 | 4.3526 | 4.8195 | 1.7700 | | 4.1892 | 3.6425 | 2.4192 | 3.6425 | H17 | 3.2465 | 2.1049 | 3.8913 | 3.2913 | 2.2232 | 3.3763 | 1.0955 | 2.6627 | 4.8195 | 4.3526 | 3.3956 | 4.3526 | 2.9683 | 2.3828 | 3.6425 | 4.1892 | | 1.7700 | 3.6425 | 2.4192 | H18 | 3.2465 | 2.1049 | 3.2913 | 3.8913 | 2.2232 | 2.6627 | 1.0955 | 3.3763 | 4.3526 | 3.3956 | 4.3526 | 4.8195 | 2.3828 | 2.9683 | 2.4192 | 3.6425 | 1.7700 | | 4.1892 | 3.6425 | H19 | 3.2465 | 2.1049 | 3.2913 | 2.2232 | 3.8913 | 2.6627 | 3.3763 | 1.0955 | 3.3956 | 4.3526 | 2.9683 | 2.3828 | 4.8195 | 4.3526 | 3.6425 | 2.4192 | 3.6425 | 4.1892 | | 1.7700 | H20 | 3.2465 | 2.1049 | 3.8913 | 2.2232 | 3.2913 | 3.3763 | 2.6627 | 1.0955 | 4.3526 | 4.8195 | 2.3828 | 2.9683 | 4.3526 | 3.3956 | 4.1892 | 3.6425 | 2.4192 | 3.6425 | 1.7700 | |
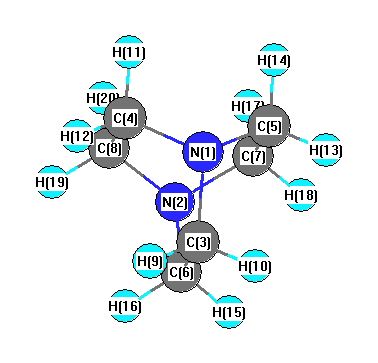 More geometry information
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.458 |
|
N1 |
C3 |
H9 |
107.977 |
| N1 |
C3 |
H10 |
107.977 |
|
N1 |
C4 |
C8 |
109.458 |
| N1 |
C4 |
H11 |
107.977 |
|
N1 |
C4 |
H12 |
107.977 |
| N1 |
C5 |
C7 |
109.458 |
|
N1 |
C5 |
H13 |
107.977 |
| N1 |
C5 |
H14 |
107.977 |
|
N2 |
C6 |
C3 |
109.458 |
| N2 |
C6 |
H15 |
107.977 |
|
N2 |
C6 |
H16 |
107.977 |
| N2 |
C7 |
C5 |
109.458 |
|
N2 |
C7 |
H17 |
107.977 |
| N2 |
C7 |
H18 |
107.977 |
|
N2 |
C8 |
C4 |
109.458 |
| N2 |
C8 |
H19 |
107.977 |
|
N2 |
C8 |
H20 |
107.977 |
| C3 |
N1 |
C4 |
109.485 |
|
C3 |
N1 |
C5 |
109.485 |
| C3 |
C6 |
H15 |
111.755 |
|
C3 |
C6 |
H16 |
111.755 |
| C4 |
N1 |
C5 |
109.485 |
|
C4 |
C8 |
H19 |
111.755 |
| C4 |
C8 |
H20 |
111.755 |
|
C5 |
C6 |
H15 |
101.246 |
| C5 |
C6 |
H16 |
150.909 |
|
C6 |
N2 |
C7 |
109.485 |
| C6 |
N2 |
C8 |
109.485 |
|
C6 |
C3 |
H9 |
111.755 |
| C6 |
C3 |
H10 |
111.755 |
|
C7 |
N2 |
C8 |
109.485 |
| C7 |
C5 |
H13 |
111.755 |
|
C7 |
C5 |
H14 |
111.755 |
| C8 |
C4 |
H11 |
111.755 |
|
C8 |
C4 |
H12 |
111.755 |
| H9 |
C3 |
H10 |
107.771 |
|
H11 |
C4 |
H12 |
107.771 |
| H13 |
C5 |
H14 |
107.771 |
|
H15 |
C6 |
H16 |
107.771 |
| H17 |
C7 |
H18 |
107.771 |
|
H19 |
C8 |
H20 |
107.771 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.401 |
|
|
|
| 2 |
N |
-0.401 |
|
|
|
| 3 |
C |
-0.145 |
|
|
|
| 4 |
C |
-0.145 |
|
|
|
| 5 |
C |
-0.145 |
|
|
|
| 6 |
C |
-0.145 |
|
|
|
| 7 |
C |
-0.145 |
|
|
|
| 8 |
C |
-0.145 |
|
|
|
| 9 |
H |
0.139 |
|
|
|
| 10 |
H |
0.139 |
|
|
|
| 11 |
H |
0.139 |
|
|
|
| 12 |
H |
0.139 |
|
|
|
| 13 |
H |
0.139 |
|
|
|
| 14 |
H |
0.139 |
|
|
|
| 15 |
H |
0.139 |
|
|
|
| 16 |
H |
0.139 |
|
|
|
| 17 |
H |
0.139 |
|
|
|
| 18 |
H |
0.139 |
|
|
|
| 19 |
H |
0.139 |
|
|
|
| 20 |
H |
0.139 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.424 |
0.000 |
0.000 |
| y |
0.000 |
-46.424 |
0.000 |
| z |
0.000 |
0.000 |
-59.008 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.292 |
0.000 |
0.000 |
| y |
0.000 |
6.292 |
0.000 |
| z |
0.000 |
0.000 |
-12.584 |
|
| Polar |
|---|
| 3z2-r2 | -25.168 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.962 |
0.000 |
0.000 |
| y |
0.000 |
10.962 |
0.000 |
| z |
0.000 |
0.000 |
9.226 |
<r2> (average value of r
2) Å
2
| <r2> |
218.056 |
| (<r2>)1/2 |
14.767 |
Jump to
S1C1
Energy calculated at B3LYP/6-31G
| | hartrees |
|---|
| Energy at 0K | -345.220475 |
| Energy at 298.15K | -345.237612 |
| HF Energy | -345.220475 |
| Nuclear repulsion energy | 420.441484 |
The energy at 298.15K was derived from the energy at 0K
and an integrated heat capacity that used the calculated vibrational frequencies.
Vibrational Frequencies calculated at B3LYP/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3092 |
2975 |
0.00 |
|
|
|
| 2 |
A1 |
3067 |
2950 |
0.00 |
|
|
|
| 3 |
A1 |
1536 |
1477 |
0.00 |
|
|
|
| 4 |
A1 |
1357 |
1305 |
0.00 |
|
|
|
| 5 |
A1 |
1283 |
1234 |
0.00 |
|
|
|
| 6 |
A1 |
1040 |
1000 |
0.00 |
|
|
|
| 7 |
A1 |
941 |
906 |
0.00 |
|
|
|
| 8 |
A1 |
793 |
763 |
0.00 |
|
|
|
| 9 |
A1 |
605 |
582 |
0.00 |
|
|
|
| 10 |
A1 |
112 |
108 |
0.00 |
|
|
|
| 11 |
A2 |
3114 |
2995 |
0.00 |
|
|
|
| 12 |
A2 |
3052 |
2936 |
122.33 |
|
|
|
| 13 |
A2 |
1532 |
1473 |
5.73 |
|
|
|
| 14 |
A2 |
1398 |
1345 |
1.75 |
|
|
|
| 15 |
A2 |
1207 |
1161 |
0.00 |
|
|
|
| 16 |
A2 |
993 |
955 |
19.29 |
|
|
|
| 17 |
A2 |
824 |
793 |
0.00 |
|
|
|
| 18 |
A2 |
709 |
682 |
68.97 |
|
|
|
| 19 |
E |
3123 |
3004 |
90.36 |
|
|
|
| 19 |
E |
3123 |
3004 |
90.34 |
|
|
|
| 20 |
E |
3097 |
2979 |
0.01 |
|
|
|
| 20 |
E |
3097 |
2979 |
0.01 |
|
|
|
| 21 |
E |
3058 |
2942 |
105.59 |
|
|
|
| 21 |
E |
3058 |
2942 |
105.63 |
|
|
|
| 22 |
E |
3049 |
2934 |
0.00 |
|
|
|
| 22 |
E |
3049 |
2934 |
0.00 |
|
|
|
| 23 |
E |
1528 |
1470 |
13.24 |
|
|
|
| 23 |
E |
1528 |
1470 |
13.23 |
|
|
|
| 24 |
E |
1521 |
1463 |
0.00 |
|
|
|
| 24 |
E |
1521 |
1463 |
0.00 |
|
|
|
| 25 |
E |
1366 |
1314 |
0.01 |
|
|
|
| 25 |
E |
1366 |
1314 |
0.01 |
|
|
|
| 26 |
E |
1364 |
1312 |
5.54 |
|
|
|
| 26 |
E |
1364 |
1312 |
5.54 |
|
|
|
| 27 |
E |
1343 |
1292 |
0.00 |
|
|
|
| 27 |
E |
1343 |
1292 |
0.00 |
|
|
|
| 28 |
E |
1338 |
1287 |
0.16 |
|
|
|
| 28 |
E |
1338 |
1287 |
0.16 |
|
|
|
| 29 |
E |
1221 |
1175 |
0.00 |
|
|
|
| 29 |
E |
1221 |
1175 |
0.00 |
|
|
|
| 30 |
E |
1056 |
1016 |
33.32 |
|
|
|
| 30 |
E |
1056 |
1016 |
33.32 |
|
|
|
| 31 |
E |
1022 |
983 |
0.00 |
|
|
|
| 31 |
E |
1022 |
983 |
0.00 |
|
|
|
| 32 |
E |
892 |
858 |
2.75 |
|
|
|
| 32 |
E |
892 |
858 |
2.75 |
|
|
|
| 33 |
E |
842 |
810 |
7.58 |
|
|
|
| 33 |
E |
842 |
810 |
7.58 |
|
|
|
| 34 |
E |
587 |
564 |
0.00 |
|
|
|
| 34 |
E |
587 |
564 |
0.00 |
|
|
|
| 35 |
E |
426 |
410 |
0.03 |
|
|
|
| 35 |
E |
426 |
410 |
0.03 |
|
|
|
| 36 |
E |
336 |
323 |
0.00 |
|
|
|
| 36 |
E |
336 |
323 |
0.00 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 40492.6 cm
-1
Scaled (by 0.962) Zero Point Vibrational Energy (zpe) 38953.9 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Geometric Data calculated at B3LYP/6-31G
Point Group is D3
Cartesians (Å)
| Atom |
x (Å) |
y (Å) |
z (Å) |
|---|
| N1 |
0.000 |
0.000 |
1.282 |
| N2 |
0.000 |
0.000 |
-1.282 |
| C3 |
-0.001 |
1.406 |
0.785 |
| C4 |
1.218 |
-0.702 |
0.785 |
| C5 |
-1.217 |
-0.704 |
0.785 |
| C6 |
0.001 |
1.406 |
-0.785 |
| C7 |
-1.218 |
-0.702 |
-0.785 |
| C8 |
1.217 |
-0.704 |
-0.785 |
| H9 |
0.882 |
1.910 |
1.193 |
| H10 |
-0.888 |
1.906 |
1.190 |
| H11 |
1.213 |
-1.718 |
1.193 |
| H12 |
2.095 |
-0.184 |
1.190 |
| H13 |
-2.095 |
-0.191 |
1.193 |
| H14 |
-1.207 |
-1.722 |
1.190 |
| H15 |
-0.882 |
1.910 |
-1.193 |
| H16 |
0.888 |
1.906 |
-1.190 |
| H17 |
-1.213 |
-1.718 |
-1.193 |
| H18 |
-2.095 |
-0.184 |
-1.190 |
| H19 |
2.095 |
-0.191 |
-1.193 |
| H20 |
1.207 |
-1.722 |
-1.190 |
Atom - Atom Distances (Å)
| |
N1 |
N2 |
C3 |
C4 |
C5 |
C6 |
C7 |
C8 |
H9 |
H10 |
H11 |
H12 |
H13 |
H14 |
H15 |
H16 |
H17 |
H18 |
H19 |
H20 |
| N1 | | 2.5641 | 1.4908 | 1.4908 | 1.4908 | 2.5000 | 2.5000 | 2.5000 | 2.1052 | 2.1048 | 2.1052 | 2.1048 | 2.1052 | 2.1048 | 3.2481 | 3.2451 | 3.2481 | 3.2451 | 3.2481 | 3.2451 |
N2 | 2.5641 | | 2.5000 | 2.5000 | 2.5000 | 1.4908 | 1.4908 | 1.4908 | 3.2481 | 3.2451 | 3.2481 | 3.2451 | 3.2481 | 3.2451 | 2.1052 | 2.1048 | 2.1052 | 2.1048 | 2.1052 | 2.1048 | C3 | 1.4908 | 2.5000 | | 2.4346 | 2.4346 | 1.5707 | 2.8961 | 2.8985 | 1.0955 | 1.0955 | 3.3764 | 2.6615 | 2.6641 | 3.3762 | 2.2233 | 2.2231 | 3.8911 | 3.2877 | 3.2950 | 3.8914 | C4 | 1.4908 | 2.5000 | 2.4346 | | 2.4346 | 2.8961 | 2.8985 | 1.5707 | 2.6641 | 3.3762 | 1.0955 | 1.0955 | 3.3764 | 2.6615 | 3.8911 | 3.2877 | 3.2950 | 3.8914 | 2.2233 | 2.2231 | C5 | 1.4908 | 2.5000 | 2.4346 | 2.4346 | | 2.8985 | 1.5707 | 2.8961 | 3.3764 | 2.6615 | 2.6641 | 3.3762 | 1.0955 | 1.0955 | 3.2950 | 3.8914 | 2.2233 | 2.2231 | 3.8911 | 3.2877 | C6 | 2.5000 | 1.4908 | 1.5707 | 2.8961 | 2.8985 | | 2.4346 | 2.4346 | 2.2233 | 2.2231 | 3.8911 | 3.2877 | 3.2950 | 3.8914 | 1.0955 | 1.0955 | 3.3764 | 2.6615 | 2.6641 | 3.3762 | C7 | 2.5000 | 1.4908 | 2.8961 | 2.8985 | 1.5707 | 2.4346 | | 2.4346 | 3.8911 | 3.2877 | 3.2950 | 3.8914 | 2.2233 | 2.2231 | 2.6641 | 3.3762 | 1.0955 | 1.0955 | 3.3764 | 2.6615 | C8 | 2.5000 | 1.4908 | 2.8985 | 1.5707 | 2.8961 | 2.4346 | 2.4346 | | 3.2950 | 3.8914 | 2.2233 | 2.2231 | 3.8911 | 3.2877 | 3.3764 | 2.6615 | 2.6641 | 3.3762 | 1.0955 | 1.0955 | H9 | 2.1052 | 3.2481 | 1.0955 | 2.6641 | 3.3764 | 2.2233 | 3.8911 | 3.2950 | | 1.7699 | 3.6431 | 2.4194 | 3.6431 | 4.1894 | 2.9670 | 2.3826 | 4.8211 | 4.3495 | 3.4026 | 4.3557 | H10 | 2.1048 | 3.2451 | 1.0955 | 3.3762 | 2.6615 | 2.2231 | 3.2877 | 3.8914 | 1.7699 | | 4.1894 | 3.6421 | 2.4194 | 3.6421 | 2.3826 | 2.9692 | 4.3495 | 3.3888 | 4.3557 | 4.8179 | H11 | 2.1052 | 3.2481 | 3.3764 | 1.0955 | 2.6641 | 3.8911 | 3.2950 | 2.2233 | 3.6431 | 4.1894 | | 1.7699 | 3.6431 | 2.4194 | 4.8211 | 4.3495 | 3.4026 | 4.3557 | 2.9670 | 2.3826 | H12 | 2.1048 | 3.2451 | 2.6615 | 1.0955 | 3.3762 | 3.2877 | 3.8914 | 2.2231 | 2.4194 | 3.6421 | 1.7699 | | 4.1894 | 3.6421 | 4.3495 | 3.3888 | 4.3557 | 4.8179 | 2.3826 | 2.9692 | H13 | 2.1052 | 3.2481 | 2.6641 | 3.3764 | 1.0955 | 3.2950 | 2.2233 | 3.8911 | 3.6431 | 2.4194 | 3.6431 | 4.1894 | | 1.7699 | 3.4026 | 4.3557 | 2.9670 | 2.3826 | 4.8211 | 4.3496 | H14 | 2.1048 | 3.2451 | 3.3762 | 2.6615 | 1.0955 | 3.8914 | 2.2231 | 3.2877 | 4.1894 | 3.6421 | 2.4194 | 3.6421 | 1.7699 | | 4.3557 | 4.8179 | 2.3826 | 2.9692 | 4.3496 | 3.3888 | H15 | 3.2481 | 2.1052 | 2.2233 | 3.8911 | 3.2950 | 1.0955 | 2.6641 | 3.3764 | 2.9670 | 2.3826 | 4.8211 | 4.3495 | 3.4026 | 4.3557 | | 1.7699 | 3.6431 | 2.4194 | 3.6431 | 4.1894 | H16 | 3.2451 | 2.1048 | 2.2231 | 3.2877 | 3.8914 | 1.0955 | 3.3762 | 2.6615 | 2.3826 | 2.9692 | 4.3495 | 3.3888 | 4.3557 | 4.8179 | 1.7699 | | 4.1894 | 3.6421 | 2.4194 | 3.6421 | H17 | 3.2481 | 2.1052 | 3.8911 | 3.2950 | 2.2233 | 3.3764 | 1.0955 | 2.6641 | 4.8211 | 4.3495 | 3.4026 | 4.3557 | 2.9670 | 2.3826 | 3.6431 | 4.1894 | | 1.7699 | 3.6431 | 2.4194 | H18 | 3.2451 | 2.1048 | 3.2877 | 3.8914 | 2.2231 | 2.6615 | 1.0955 | 3.3762 | 4.3495 | 3.3888 | 4.3557 | 4.8179 | 2.3826 | 2.9692 | 2.4194 | 3.6421 | 1.7699 | | 4.1894 | 3.6421 | H19 | 3.2481 | 2.1052 | 3.2950 | 2.2233 | 3.8911 | 2.6641 | 3.3764 | 1.0955 | 3.4026 | 4.3557 | 2.9670 | 2.3826 | 4.8211 | 4.3496 | 3.6431 | 2.4194 | 3.6431 | 4.1894 | | 1.7699 | H20 | 3.2451 | 2.1048 | 3.8914 | 2.2231 | 3.2877 | 3.3762 | 2.6615 | 1.0955 | 4.3557 | 4.8179 | 2.3826 | 2.9692 | 4.3496 | 3.3888 | 4.1894 | 3.6421 | 2.4194 | 3.6421 | 1.7699 | |
More geometry information
Calculated Bond Angles
| atom1 |
atom2 |
atom3 |
angle |
|
atom1 |
atom2 |
atom3 |
angle |
| N1 |
C3 |
C6 |
109.462 |
|
N1 |
C3 |
H9 |
108.002 |
| N1 |
C3 |
H10 |
107.966 |
|
N1 |
C4 |
C8 |
109.462 |
| N1 |
C4 |
H11 |
108.002 |
|
N1 |
C4 |
H12 |
107.966 |
| N1 |
C5 |
C7 |
109.462 |
|
N1 |
C5 |
H13 |
108.002 |
| N1 |
C5 |
H14 |
107.966 |
|
N2 |
C6 |
C3 |
109.462 |
| N2 |
C6 |
H15 |
108.002 |
|
N2 |
C6 |
H16 |
107.966 |
| N2 |
C7 |
C5 |
109.462 |
|
N2 |
C7 |
H17 |
108.002 |
| N2 |
C7 |
H18 |
107.966 |
|
N2 |
C8 |
C4 |
109.462 |
| N2 |
C8 |
H19 |
108.002 |
|
N2 |
C8 |
H20 |
107.966 |
| C3 |
N1 |
C4 |
109.481 |
|
C3 |
N1 |
C5 |
109.481 |
| C3 |
C6 |
H15 |
111.757 |
|
C3 |
C6 |
H16 |
111.745 |
| C4 |
N1 |
C5 |
109.481 |
|
C4 |
C8 |
H19 |
111.757 |
| C4 |
C8 |
H20 |
111.745 |
|
C5 |
C6 |
H15 |
101.405 |
| C5 |
C6 |
H16 |
150.758 |
|
C6 |
N2 |
C7 |
109.481 |
| C6 |
N2 |
C8 |
109.481 |
|
C6 |
C3 |
H9 |
111.757 |
| C6 |
C3 |
H10 |
111.745 |
|
C7 |
N2 |
C8 |
109.481 |
| C7 |
C5 |
H13 |
111.757 |
|
C7 |
C5 |
H14 |
111.745 |
| C8 |
C4 |
H11 |
111.757 |
|
C8 |
C4 |
H12 |
111.745 |
| H9 |
C3 |
H10 |
107.763 |
|
H11 |
C4 |
H12 |
107.763 |
| H13 |
C5 |
H14 |
107.763 |
|
H15 |
C6 |
H16 |
107.763 |
| H17 |
C7 |
H18 |
107.763 |
|
H19 |
C8 |
H20 |
107.763 |
Electronic energy levels
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
N |
-0.401 |
|
|
|
| 2 |
N |
-0.401 |
|
|
|
| 3 |
C |
-0.145 |
|
|
|
| 4 |
C |
-0.145 |
|
|
|
| 5 |
C |
-0.145 |
|
|
|
| 6 |
C |
-0.145 |
|
|
|
| 7 |
C |
-0.145 |
|
|
|
| 8 |
C |
-0.145 |
|
|
|
| 9 |
H |
0.139 |
|
|
|
| 10 |
H |
0.139 |
|
|
|
| 11 |
H |
0.139 |
|
|
|
| 12 |
H |
0.139 |
|
|
|
| 13 |
H |
0.139 |
|
|
|
| 14 |
H |
0.139 |
|
|
|
| 15 |
H |
0.139 |
|
|
|
| 16 |
H |
0.139 |
|
|
|
| 17 |
H |
0.139 |
|
|
|
| 18 |
H |
0.139 |
|
|
|
| 19 |
H |
0.139 |
|
|
|
| 20 |
H |
0.139 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.000 |
0.000 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.423 |
0.000 |
0.000 |
| y |
0.000 |
-46.423 |
0.000 |
| z |
0.000 |
0.000 |
-59.010 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
6.293 |
0.000 |
0.000 |
| y |
0.000 |
6.293 |
0.000 |
| z |
0.000 |
0.000 |
-12.587 |
|
| Polar |
|---|
| 3z2-r2 | -25.174 |
|---|
| x2-y2 | 0.000 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
10.962 |
0.000 |
0.000 |
| y |
0.000 |
10.962 |
0.000 |
| z |
0.000 |
0.000 |
9.226 |
<r2> (average value of r
2) Å
2
| <r2> |
218.061 |
| (<r2>)1/2 |
14.767 |