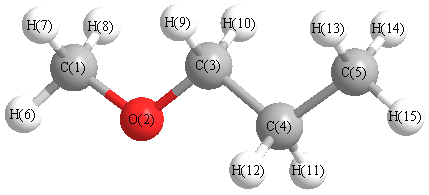
Vibrational Frequencies calculated at B3LYP/6-31G
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A' |
3158 |
3038 |
29.06 |
|
|
|
| 2 |
A' |
3122 |
3003 |
40.04 |
|
|
|
| 3 |
A' |
3056 |
2940 |
25.96 |
|
|
|
| 4 |
A' |
3039 |
2923 |
31.81 |
|
|
|
| 5 |
A' |
2997 |
2884 |
71.09 |
|
|
|
| 6 |
A' |
2971 |
2858 |
38.39 |
|
|
|
| 7 |
A' |
1570 |
1510 |
2.59 |
|
|
|
| 8 |
A' |
1553 |
1494 |
5.21 |
|
|
|
| 9 |
A' |
1545 |
1486 |
2.43 |
|
|
|
| 10 |
A' |
1540 |
1482 |
10.34 |
|
|
|
| 11 |
A' |
1501 |
1444 |
0.42 |
|
|
|
| 12 |
A' |
1464 |
1408 |
9.26 |
|
|
|
| 13 |
A' |
1437 |
1382 |
13.18 |
|
|
|
| 14 |
A' |
1371 |
1319 |
3.23 |
|
|
|
| 15 |
A' |
1218 |
1171 |
14.50 |
|
|
|
| 16 |
A' |
1153 |
1110 |
9.07 |
|
|
|
| 17 |
A' |
1096 |
1054 |
130.59 |
|
|
|
| 18 |
A' |
1065 |
1024 |
0.86 |
|
|
|
| 19 |
A' |
939 |
903 |
2.30 |
|
|
|
| 20 |
A' |
907 |
873 |
21.23 |
|
|
|
| 21 |
A' |
429 |
412 |
1.03 |
|
|
|
| 22 |
A' |
406 |
391 |
3.45 |
|
|
|
| 23 |
A' |
190 |
183 |
2.38 |
|
|
|
| 24 |
A" |
3117 |
2998 |
80.42 |
|
|
|
| 25 |
A" |
3089 |
2972 |
0.67 |
|
|
|
| 26 |
A" |
3051 |
2935 |
73.69 |
|
|
|
| 27 |
A" |
3005 |
2891 |
60.26 |
|
|
|
| 28 |
A" |
1547 |
1488 |
8.30 |
|
|
|
| 29 |
A" |
1526 |
1468 |
7.72 |
|
|
|
| 30 |
A" |
1339 |
1288 |
0.00 |
|
|
|
| 31 |
A" |
1286 |
1237 |
0.91 |
|
|
|
| 32 |
A" |
1207 |
1161 |
4.09 |
|
|
|
| 33 |
A" |
1162 |
1118 |
0.40 |
|
|
|
| 34 |
A" |
931 |
896 |
2.07 |
|
|
|
| 35 |
A" |
795 |
765 |
2.61 |
|
|
|
| 36 |
A" |
239 |
230 |
0.03 |
|
|
|
| 37 |
A" |
222 |
214 |
4.77 |
|
|
|
| 38 |
A" |
113 |
109 |
4.09 |
|
|
|
| 39 |
A" |
99 |
95 |
1.54 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 30226.4 cm
-1
Scaled (by 0.962) Zero Point Vibrational Energy (zpe) 29077.8 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B3LYP/6-31G
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.154 |
|
|
|
| 2 |
O |
-0.525 |
|
|
|
| 3 |
C |
0.006 |
|
|
|
| 4 |
C |
-0.242 |
|
|
|
| 5 |
C |
-0.417 |
|
|
|
| 6 |
H |
0.157 |
|
|
|
| 7 |
H |
0.126 |
|
|
|
| 8 |
H |
0.126 |
|
|
|
| 9 |
H |
0.115 |
|
|
|
| 10 |
H |
0.115 |
|
|
|
| 11 |
H |
0.144 |
|
|
|
| 12 |
H |
0.144 |
|
|
|
| 13 |
H |
0.133 |
|
|
|
| 14 |
H |
0.133 |
|
|
|
| 15 |
H |
0.139 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.146 |
1.466 |
0.000 |
1.473 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-29.984 |
-2.509 |
0.000 |
| y |
-2.509 |
-33.752 |
0.000 |
| z |
0.000 |
0.000 |
-33.140 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
3.462 |
-2.509 |
0.000 |
| y |
-2.509 |
-2.190 |
0.000 |
| z |
0.000 |
0.000 |
-1.271 |
|
| Polar |
|---|
| 3z2-r2 | -2.543 |
|---|
| x2-y2 | 3.768 |
|---|
| xy | -2.509 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
8.584 |
-0.251 |
0.000 |
| y |
-0.251 |
6.427 |
0.000 |
| z |
0.000 |
0.000 |
6.261 |
<r2> (average value of r
2) Å
2
| <r2> |
185.399 |
| (<r2>)1/2 |
13.616 |