Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A |
3083 |
3022 |
27.64 |
|
|
|
| 2 |
A |
3081 |
3021 |
41.38 |
|
|
|
| 3 |
A |
3078 |
3018 |
34.05 |
|
|
|
| 4 |
A |
3066 |
3006 |
38.50 |
|
|
|
| 5 |
A |
3062 |
3002 |
68.34 |
|
|
|
| 6 |
A |
3055 |
2995 |
0.69 |
|
|
|
| 7 |
A |
2999 |
2940 |
41.47 |
|
|
|
| 8 |
A |
2996 |
2938 |
33.86 |
|
|
|
| 9 |
A |
2991 |
2932 |
17.04 |
|
|
|
| 10 |
A |
2987 |
2928 |
19.69 |
|
|
|
| 11 |
A |
2971 |
2913 |
4.80 |
|
|
|
| 12 |
A |
2637 |
2585 |
35.14 |
|
|
|
| 13 |
A |
1526 |
1496 |
2.02 |
|
|
|
| 14 |
A |
1508 |
1478 |
18.05 |
|
|
|
| 15 |
A |
1507 |
1478 |
2.55 |
|
|
|
| 16 |
A |
1496 |
1466 |
2.96 |
|
|
|
| 17 |
A |
1493 |
1464 |
2.33 |
|
|
|
| 18 |
A |
1487 |
1458 |
0.80 |
|
|
|
| 19 |
A |
1425 |
1397 |
4.20 |
|
|
|
| 20 |
A |
1413 |
1385 |
5.94 |
|
|
|
| 21 |
A |
1402 |
1374 |
1.86 |
|
|
|
| 22 |
A |
1372 |
1345 |
0.40 |
|
|
|
| 23 |
A |
1338 |
1312 |
0.92 |
|
|
|
| 24 |
A |
1313 |
1287 |
4.72 |
|
|
|
| 25 |
A |
1245 |
1220 |
33.84 |
|
|
|
| 26 |
A |
1186 |
1162 |
2.55 |
|
|
|
| 27 |
A |
1168 |
1145 |
9.22 |
|
|
|
| 28 |
A |
1138 |
1116 |
0.61 |
|
|
|
| 29 |
A |
1077 |
1056 |
2.25 |
|
|
|
| 30 |
A |
1029 |
1009 |
9.04 |
|
|
|
| 31 |
A |
981 |
962 |
2.49 |
|
|
|
| 32 |
A |
962 |
943 |
0.41 |
|
|
|
| 33 |
A |
929 |
911 |
0.81 |
|
|
|
| 34 |
A |
911 |
894 |
2.30 |
|
|
|
| 35 |
A |
890 |
872 |
3.14 |
|
|
|
| 36 |
A |
776 |
761 |
3.10 |
|
|
|
| 37 |
A |
668 |
655 |
4.08 |
|
|
|
| 38 |
A |
472 |
463 |
0.57 |
|
|
|
| 39 |
A |
409 |
401 |
0.65 |
|
|
|
| 40 |
A |
374 |
366 |
0.30 |
|
|
|
| 41 |
A |
368 |
360 |
0.29 |
|
|
|
| 42 |
A |
329 |
323 |
1.48 |
|
|
|
| 43 |
A |
256 |
251 |
1.72 |
|
|
|
| 44 |
A |
241 |
236 |
2.35 |
|
|
|
| 45 |
A |
227 |
223 |
0.09 |
|
|
|
| 46 |
A |
196 |
192 |
3.02 |
|
|
|
| 47 |
A |
181 |
177 |
14.54 |
|
|
|
| 48 |
A |
55 |
54 |
1.29 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34675.1 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 33995.4 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
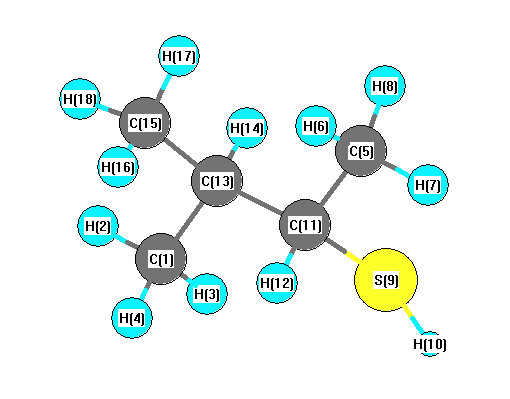
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.465 |
|
|
|
| 2 |
H |
0.147 |
|
|
|
| 3 |
H |
0.176 |
|
|
|
| 4 |
H |
0.147 |
|
|
|
| 5 |
C |
-0.458 |
|
|
|
| 6 |
H |
0.159 |
|
|
|
| 7 |
H |
0.150 |
|
|
|
| 8 |
H |
0.167 |
|
|
|
| 9 |
S |
-0.104 |
|
|
|
| 10 |
H |
0.087 |
|
|
|
| 11 |
C |
-0.221 |
|
|
|
| 12 |
H |
0.164 |
|
|
|
| 13 |
C |
-0.082 |
|
|
|
| 14 |
H |
0.146 |
|
|
|
| 15 |
C |
-0.463 |
|
|
|
| 16 |
H |
0.146 |
|
|
|
| 17 |
H |
0.154 |
|
|
|
| 18 |
H |
0.153 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
-0.974 |
1.422 |
0.586 |
1.821 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-46.463 |
2.806 |
1.649 |
| y |
2.806 |
-47.701 |
0.717 |
| z |
1.649 |
0.717 |
-47.726 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
1.250 |
2.806 |
1.649 |
| y |
2.806 |
-0.607 |
0.717 |
| z |
1.649 |
0.717 |
-0.644 |
|
| Polar |
|---|
| 3z2-r2 | -1.288 |
|---|
| x2-y2 | 1.238 |
|---|
| xy | 2.806 |
|---|
| xz | 1.649 |
|---|
| yz | 0.717 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
12.303 |
-0.264 |
0.057 |
| y |
-0.264 |
10.782 |
0.236 |
| z |
0.057 |
0.236 |
8.560 |
<r2> (average value of r
2) Å
2
| <r2> |
252.158 |
| (<r2>)1/2 |
15.879 |