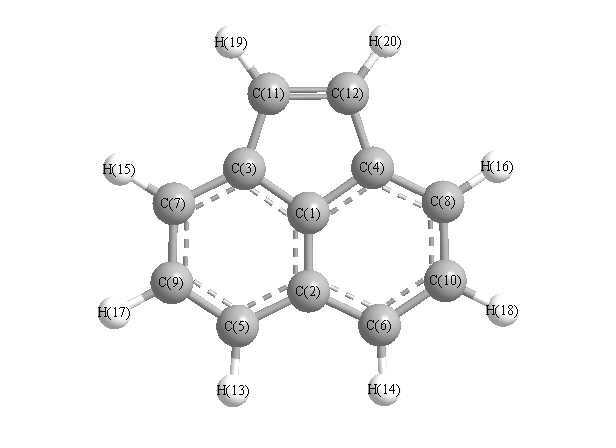
Vibrational Frequencies calculated at B97D3/6-31G*
| Mode Number |
Symmetry |
Frequency
(cm-1) |
Scaled Frequency
(cm-1) |
IR Intensities
(km mol-1) |
Raman Act
(Å4/u) |
Dep P |
Dep U |
|---|
| 1 |
A1 |
3189 |
3127 |
29.19 |
|
|
|
| 2 |
A1 |
3145 |
3083 |
21.07 |
|
|
|
| 3 |
A1 |
3130 |
3069 |
39.34 |
|
|
|
| 4 |
A1 |
3120 |
3059 |
0.68 |
|
|
|
| 5 |
A1 |
1627 |
1596 |
0.31 |
|
|
|
| 6 |
A1 |
1609 |
1577 |
0.01 |
|
|
|
| 7 |
A1 |
1512 |
1482 |
10.15 |
|
|
|
| 8 |
A1 |
1453 |
1424 |
33.44 |
|
|
|
| 9 |
A1 |
1432 |
1403 |
0.92 |
|
|
|
| 10 |
A1 |
1370 |
1343 |
1.01 |
|
|
|
| 11 |
A1 |
1256 |
1232 |
0.43 |
|
|
|
| 12 |
A1 |
1192 |
1169 |
3.66 |
|
|
|
| 13 |
A1 |
1090 |
1069 |
12.48 |
|
|
|
| 14 |
A1 |
1046 |
1025 |
1.67 |
|
|
|
| 15 |
A1 |
1015 |
996 |
1.64 |
|
|
|
| 16 |
A1 |
805 |
790 |
0.34 |
|
|
|
| 17 |
A1 |
667 |
654 |
0.27 |
|
|
|
| 18 |
A1 |
554 |
543 |
2.94 |
|
|
|
| 19 |
A1 |
416 |
408 |
1.57 |
|
|
|
| 20 |
A2 |
926 |
908 |
0.00 |
|
|
|
| 21 |
A2 |
887 |
869 |
0.00 |
|
|
|
| 22 |
A2 |
872 |
855 |
0.00 |
|
|
|
| 23 |
A2 |
740 |
726 |
0.00 |
|
|
|
| 24 |
A2 |
660 |
647 |
0.00 |
|
|
|
| 25 |
A2 |
565 |
554 |
0.00 |
|
|
|
| 26 |
A2 |
365 |
358 |
0.00 |
|
|
|
| 27 |
A2 |
206 |
202 |
0.00 |
|
|
|
| 28 |
B1 |
936 |
918 |
1.47 |
|
|
|
| 29 |
B1 |
890 |
873 |
2.09 |
|
|
|
| 30 |
B1 |
826 |
810 |
40.59 |
|
|
|
| 31 |
B1 |
764 |
749 |
36.26 |
|
|
|
| 32 |
B1 |
717 |
703 |
35.41 |
|
|
|
| 33 |
B1 |
602 |
590 |
0.11 |
|
|
|
| 34 |
B1 |
447 |
438 |
0.01 |
|
|
|
| 35 |
B1 |
222 |
217 |
2.86 |
|
|
|
| 36 |
B1 |
155 |
152 |
4.40 |
|
|
|
| 37 |
B2 |
3169 |
3107 |
4.38 |
|
|
|
| 38 |
B2 |
3144 |
3082 |
76.61 |
|
|
|
| 39 |
B2 |
3129 |
3068 |
10.13 |
|
|
|
| 40 |
B2 |
3119 |
3058 |
0.04 |
|
|
|
| 41 |
B2 |
1631 |
1599 |
2.58 |
|
|
|
| 42 |
B2 |
1497 |
1467 |
8.81 |
|
|
|
| 43 |
B2 |
1470 |
1441 |
19.24 |
|
|
|
| 44 |
B2 |
1414 |
1386 |
2.84 |
|
|
|
| 45 |
B2 |
1322 |
1296 |
1.21 |
|
|
|
| 46 |
B2 |
1234 |
1210 |
4.91 |
|
|
|
| 47 |
B2 |
1211 |
1187 |
0.77 |
|
|
|
| 48 |
B2 |
1169 |
1147 |
2.45 |
|
|
|
| 49 |
B2 |
1097 |
1076 |
1.91 |
|
|
|
| 50 |
B2 |
1028 |
1007 |
0.13 |
|
|
|
| 51 |
B2 |
867 |
850 |
3.24 |
|
|
|
| 52 |
B2 |
687 |
673 |
0.00 |
|
|
|
| 53 |
B2 |
511 |
501 |
1.51 |
|
|
|
| 54 |
B2 |
464 |
455 |
1.20 |
|
|
|
Unscaled Zero Point Vibrational Energy (zpe) 34285.7 cm
-1
Scaled (by 0.9804) Zero Point Vibrational Energy (zpe) 33613.7 cm
-1
See section
III.C.1 List or set vibrational scaling factors
to change the scale factors used here.
See section
III.C.2
Calculate a vibrational scaling factor for a given set of molecules
to determine the least squares best scaling factor.
Charges, Dipole, Quadrupole and Polarizability
Charges from optimized geometry at B97D3/6-31G*
Charges (e)
| Number |
Element |
Mulliken |
CHELPG |
AIM |
ESP |
| 1 |
C |
-0.062 |
|
|
|
| 2 |
C |
0.199 |
|
|
|
| 3 |
C |
-0.208 |
|
|
|
| 4 |
C |
-0.208 |
|
|
|
| 5 |
C |
0.139 |
|
|
|
| 6 |
C |
0.139 |
|
|
|
| 7 |
C |
-0.133 |
|
|
|
| 8 |
C |
-0.133 |
|
|
|
| 9 |
C |
-0.222 |
|
|
|
| 10 |
C |
-0.222 |
|
|
|
| 11 |
C |
-0.192 |
|
|
|
| 12 |
C |
-0.192 |
|
|
|
| 13 |
H |
0.135 |
|
|
|
| 14 |
H |
0.135 |
|
|
|
| 15 |
H |
0.137 |
|
|
|
| 16 |
H |
0.137 |
|
|
|
| 17 |
H |
0.139 |
|
|
|
| 18 |
H |
0.139 |
|
|
|
| 19 |
H |
0.136 |
|
|
|
| 20 |
H |
0.136 |
|
|
|
Electric dipole moments
Electric dipole components in Debye
(What's a Debye? See section
VII.A.3)
| |
x |
y |
z |
Total |
| |
0.000 |
0.000 |
0.380 |
0.380 |
| CHELPG |
|
|
|
|
| AIM |
|
|
|
|
| ESP |
|
|
|
|
Electric Quadrupole moment
Quadrupole components in D Å
| Primitive |
|---|
| | x | y | z |
|---|
| x |
-72.813 |
0.000 |
0.000 |
| y |
0.000 |
-58.335 |
0.000 |
| z |
0.000 |
0.000 |
-60.116 |
|
| Traceless |
|---|
| | x | y | z |
|---|
| x |
-13.587 |
0.000 |
0.000 |
| y |
0.000 |
8.129 |
0.000 |
| z |
0.000 |
0.000 |
5.458 |
|
| Polar |
|---|
| 3z2-r2 | 10.916 |
|---|
| x2-y2 | -14.477 |
|---|
| xy | 0.000 |
|---|
| xz | 0.000 |
|---|
| yz | 0.000 |
|---|
|
Polarizabilities
Components of the polarizability tensor.
Units are
Å
3 (Angstrom cubed)
Change units.
| |
x |
y |
z |
| x |
5.705 |
0.000 |
0.000 |
| y |
0.000 |
25.935 |
0.000 |
| z |
0.000 |
0.000 |
22.602 |
<r2> (average value of r
2) Å
2
| <r2> |
466.008 |
| (<r2>)1/2 |
21.587 |